BugSeq’s Approach to AMR¶
Introduction¶
Antimicrobial resistance (AMR) poses a major threat to human and animal health. In 2019 alone, there were an estimated 1.27 million deaths attributable to AMR, and that number has likely grown since. For each person with an infection, AMR reduces the likelihood that they are treated with effective antimicrobials and recover from infection. Faster, more accurate detection of AMR from clinical, biothreat and surveillance samples is of paramount importance to curb the global burden of disease attributable to AMR. Prediction of phenotypic AMR using DNA (and RNA!) sequencing is a promising avenue to achieve this goal. At BugSeq, we have focused on combating AMR since our inception; to date, we have predicted AMR for hundreds of thousands of genomes. We often get questions about how our AMR analysis works; below, we detail our goals, latest approaches and updated thinking to bioinformatic analysis for AMR prediction.
Goals¶
At BugSeq, we are working to enable labs and their stakeholders, including patients, clinicians, epidemiologists and policy makers, to combat AMR using DNA/RNA sequencing. We seek:
- To accurately predict phenotype for clinically relevant antimicrobials following widely accepted standards (e.g. CLSI and EUCAST). We measure our success with standardized metrics, including categorical agreement, very major error rate, major error rate and minor error rate.
- To ensure our predictions are easily interpretable (more on this later!) and provide stakeholders with the necessary information to make infection control, policy and (in the future) treatment decisions.
- To enable prediction for every organism within a sample, whether that is a bacterial isolate or a polymicrobial clinical sample. We believe in a future where metagenomic diagnostics are used routinely in infectious disease care; to achieve this future, it will be vital to disambiguate AMR for each organism in a sample, including plasmids and other mobile genetic elements.
- To integrate and embed AMR prediction within a comprehensive bioinformatic analysis and platform. Clinical and public health laboratories need a solution from raw reads to reports, and AMR prediction is only one component of this puzzle. Beyond the analysis, many factors (e.g. support, quality assurance, data security, etc.) are required to translate bioinformatics into clinical, public health and other reference laboratories; see our recent blog post on versioning for a good example of this.
How we have tackled AMR to-date¶
Our team and collaborators have published previously on approaches we have taken to predict phenotypic AMR, and performance of those approaches. We have covered a range of sequencing platforms (Nanopore and Illumina), kingdoms (bacterial to viral), organisms (Shigella sonnei to Neisseria gonorrhoeae) and sample types (isolates to metagenomic). We have demonstrated detection of both genes and variants (single nucleotide or insertions and deletions) conferring resistance, and innovated across these areas (e.g. open source contribution to ResFinder). We have had the privilege of serving these results to CDC and other government agencies across the world, and our users have published on their experiences too.
In general, these approaches have performed highly accurately and led on performance benchmarks. Recently, we have been working to enable even greater gains in accuracy and usability. First, some more background on how we are enabling clinically-focused AMR from sequencing data.
In 2022, we and collaborators at University of British Columbia, British Columbia Centre for Disease Control and Vancouver Coastal Health published BugSplit, a method to taxonomically bin sequences. BugSplit innovated separating sequences from a de novo assembly by taxonomic origin, enabling characterization of each organism within a sample. BugSplit has stood the test of time and is still a fundamental driver of BugSeq’s accurate isolate identification and metagenomic classification: to our knowledge, there has yet to be a method published which is more accurate for taxonomic binning of sequences. In brief, BugSplit functions by aligning contigs against a reference sequence database and taking average nucleotide identity and sequence coverage into account to make a taxonomic classification. In our publication, we demonstrated that we could isolate complete and pure pathogen genomes from metagenomic data of blood and urine samples, and then predict AMR for each organism in the sample based on their respective genome.
Since then, we have significantly improved the accuracy and speed of BugSplit, with upgrades such as better plasmid detection compared with the original PlasmidFinder, utilization of the assembly graph for label refinement, and alignment against a highly curated reference sequence database (BugRef; read more in our publication).
In our original BugSplit publication, we used ResFinder+PointFinder to predict phenotypic resistance. We quickly abandoned that approach due to significant limitations, and built a better, homegrown solution:
- Systematic reporting of select drugs with CLSI breakpoints
- Handling of frameshifts in old Nanopore data (or newer data basecalled in “fast mode”, which is still popular!)
- Up to date reference database (ResFinder only has 529 blaOXAs at the time of writing, while there are now over 1000)
- Use of translated sequence to identify gene alleles (other tools get this wrong by using nucleic acid sequence)
Another power of BugSplit is that we identify the species of origin for each sequence. The scientific evidence base is clear that taxonomy has large impacts on antimicrobial resistance:
- Certain proteins are intrinsic in a species and therefore only causes resistance when found on a plasmid in a different species (e.g. oqxA/B)
- Mutations are usually species-specific
- Even if the same protein is two different species, it may have differing effects due to other factors. An interesting example of this phenomenon is blaOXA-1, which in some publications causes cefepime resistance in Pseudomonas aeruginosa but not Escherichia coli. This discordance is thought to be due to differences in intracellular drug concentration.
With that background, let us move on to some recent upgrades to our AMR prediction.
What’s new: Reporting¶
BugSeq’s AMR reports have undergone many rounds of iteration with clinical and public health labs to enable easy interpretation from the genomic data.
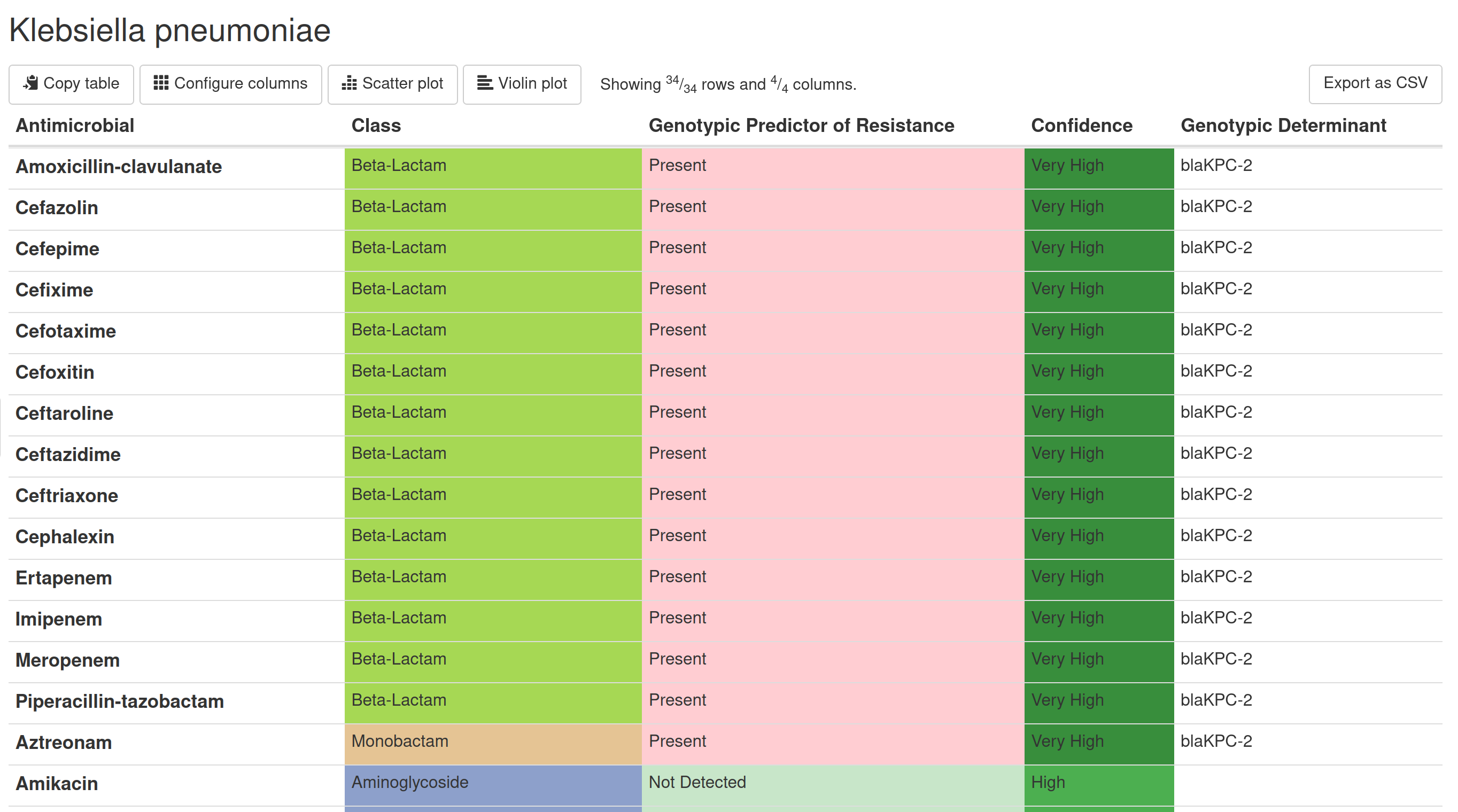
Starting at the left, we can see that BugSeq analyzes AMR by individual drugs. This reporting differentiates us from AMRFinder and CARD, who report resistance by class of drug only. However, as you can see from the above figure, resistance to one beta-lactam does not mean that there is resistance to all beta-lactams! This observation is obvious to any microbiologist but inconsistent with reporting frameworks from popular open source tools.
In the middle, we can see whether there is a Genotypic Predictor of Resistance present for individual drugs. BugSeq uses a highly curated database to detect genotypic determinants. Our team has collected determinants from online databases and is constantly scanning the literature for new determinants. We have spent countless hours linking genotype to individual phenotype. We have also built databases from scratch for features that are not captured in any public tool/database but are vital for accurate AMR prediction, such as the presence of insertion sequences or functional knockout of proteins. Protein knockout is highly predictive of phenotypic resistance in certain species, such as oprD in Pseudomonas aeruginosa and ompK36 in Klebsiella pneumoniae.
Coming soon will be a change of column title from “Genotypic Predictor of Resistance” to “Predicted CLSI Phenotype”. If we assume that “Present” translates into predicted phenotypic resistance and “Not Detected” into phenotypic susceptibility, the upcoming change will enable reports to include additional phenotypic predictions, including intermediate and susceptible dose dependent categories. Enabling these categories will further reduce very major and major errors while increasing categorical agreement. The “Genotypic Determinant” column will remain and show determinants for the predicted phenotype. As always, we will be rolling this out to our users in stages to ensure we get this right.
Last from right, we can see Confidence. We want our users to understand how confident we are in our prediction so they can make informed decisions, whether that is surveillance, infection control or (eventually) treatment. Confidence is described in our docs and calculated differently if there is a genotypic predictor present or absent. In version 5.2 (May 2024), we added confidence for the absence of AMR - this was a highly requested feature and is vital for interpretation of clinical metagenomic data, where only partial genomes for each organism may be recovered. If the pathogen is at very low abundance, it may be impossible to see enough of the organism’s genome to accurately predict AMR!
Conclusion¶
AMR continues to exact a significant global burden of disease, and this is only expected to grow. In parallel, we expect the use of genomics to understand and combat AMR to be ever more important. Accurate, high quality bioinformatic analysis paired with curated, up-to-date databases is of paramount importance to combat AMR: this is our core at BugSeq. We are investing heavily in a future where genomics can be used for clinical treatment decisions to reduce the global burden of AMR. What does this mean for BugSeq users?
- Accurate phenotypic AMR prediction for every organism in a sample
- AMR prediction meeting accepted standards for use in mission-critical settings like clinical and public health labs
- Reliable, rapid results with 24/7 support
In a future post, we will detail some of the exciting work on the underlying AMR prediction engine and recent performance estimates.
Interested in learning more?¶
- Contact Us: contact@bugseq.com
- Request a Quote: bugseq.com/quote
- Publications: bugseq.com/publications