H1 2025 Platform Update: AI-Driven Insights, Phenotypic AMR Prediction, Advanced Fungal Genomics, and Next-Generation Surveillance Tools from BugSeq¶
The first half of 2025 marked a significant leap forward for the BugSeq platform, delivering powerful new capabilities to tackle the most pressing challenges in infectious disease. We’re introducing AI-powered report summaries to accelerate interpretation, leading accuracy AMR phenotype prediction (coming in a separate blog post), advanced fungal diagnostics to combat emerging pathogens, and next-generation surveillance tools that fuse genomic and epidemiological data. These updates are engineered to empower laboratories with deeper insights, greater accuracy, and faster time to action.
Introduction: Meeting the Evolving Challenges of Modern Microbiology¶
Clinical and public health microbiology face ongoing challenges from emerging pathogens and antimicrobial resistance. For laboratories to use next-generation sequencing (NGS) effectively, they need bioinformatic solutions that manage the underlying complexity and ensure every result is accurate, reliable and reproducible; at BugSeq, this is our core mission. The BugSeq platform enhancements released in the first half of 2025 are a response to the needs of clinical, public health, and research laboratories. This post details new features for high-resolution pathogen typing, mycology and data reporting.
Enhanced Data Interpretation and Visualization¶
Metadata Overlay for Enhanced Outbreak Visualization¶
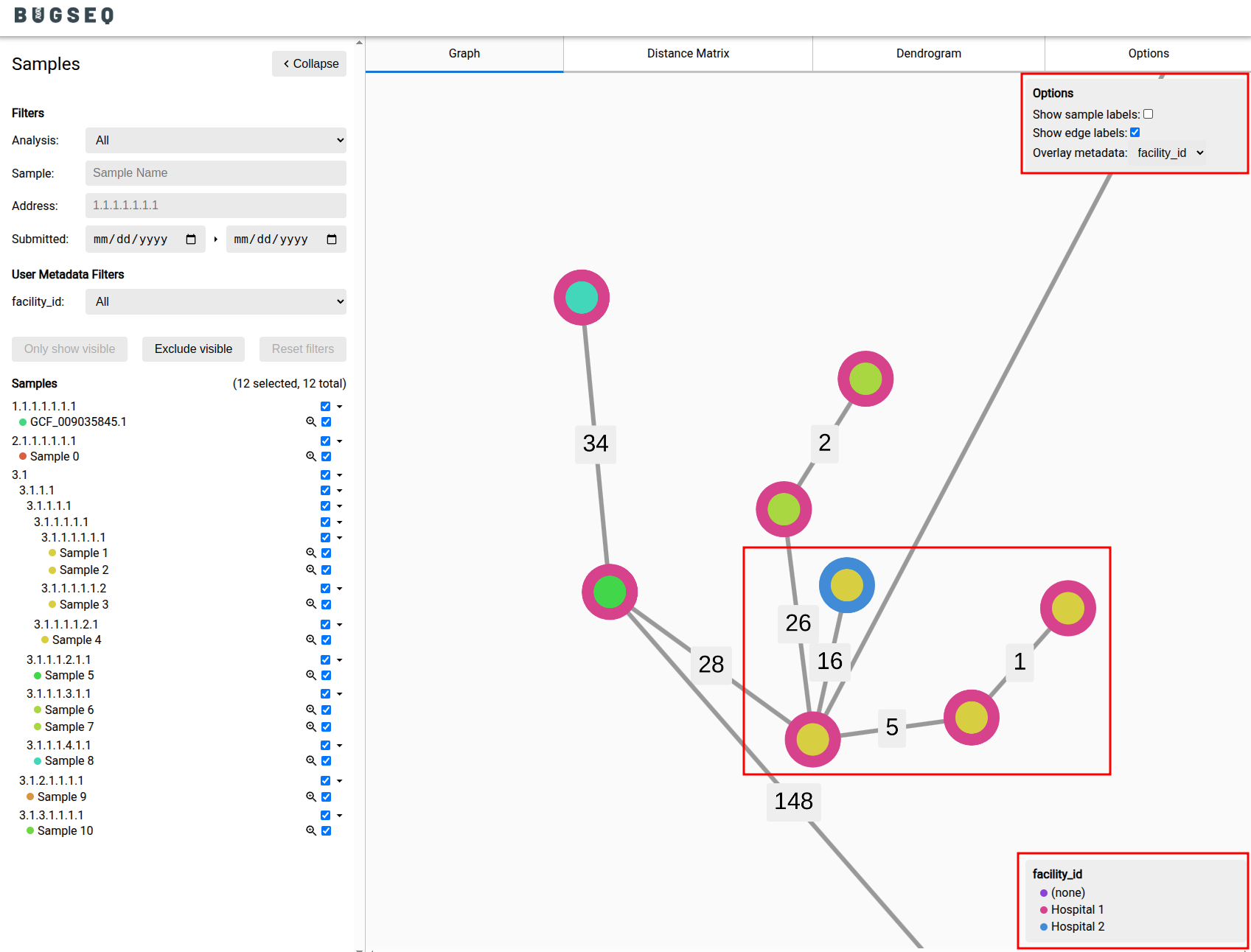

Effective outbreak investigation requires the fusion of genomic data with epidemiological context. The outbreak visualizer has been upgraded to allow users to overlay categorical metadata—such as collection facility or hospital ward—directly onto the visualizations; transforming the outbreak analysis module from a purely genomic tool into an integrated epidemiological workspace.
For example, an infection control practitioner can now color-code the tree by ward to visualize a potential transmission chain or by facility to investigate a regional outbreak.
AI-Powered Report Summaries¶

One of the most significant barriers to the widespread adoption of next-generation sequencing (NGS) in microbiology is the challenge of data interpretation. The H1 2025 updates include features designed to improve interpretation, visualization, and quality control.
- Version 5.7 of the platform introduced AI-driven interpretations within its interactive reports.
- This feature provides an automated, plain-language summary of the key findings in a sample to reduce interpretation time.
- In parallel, BugSeq has been busy implementing data interpretation tools for clinical metagenomic data with BARDA and HaDEA, as detailed in our press releases.
See example AI summaries in demo reports on our Solutions page.
Genome Coverage Visualization¶
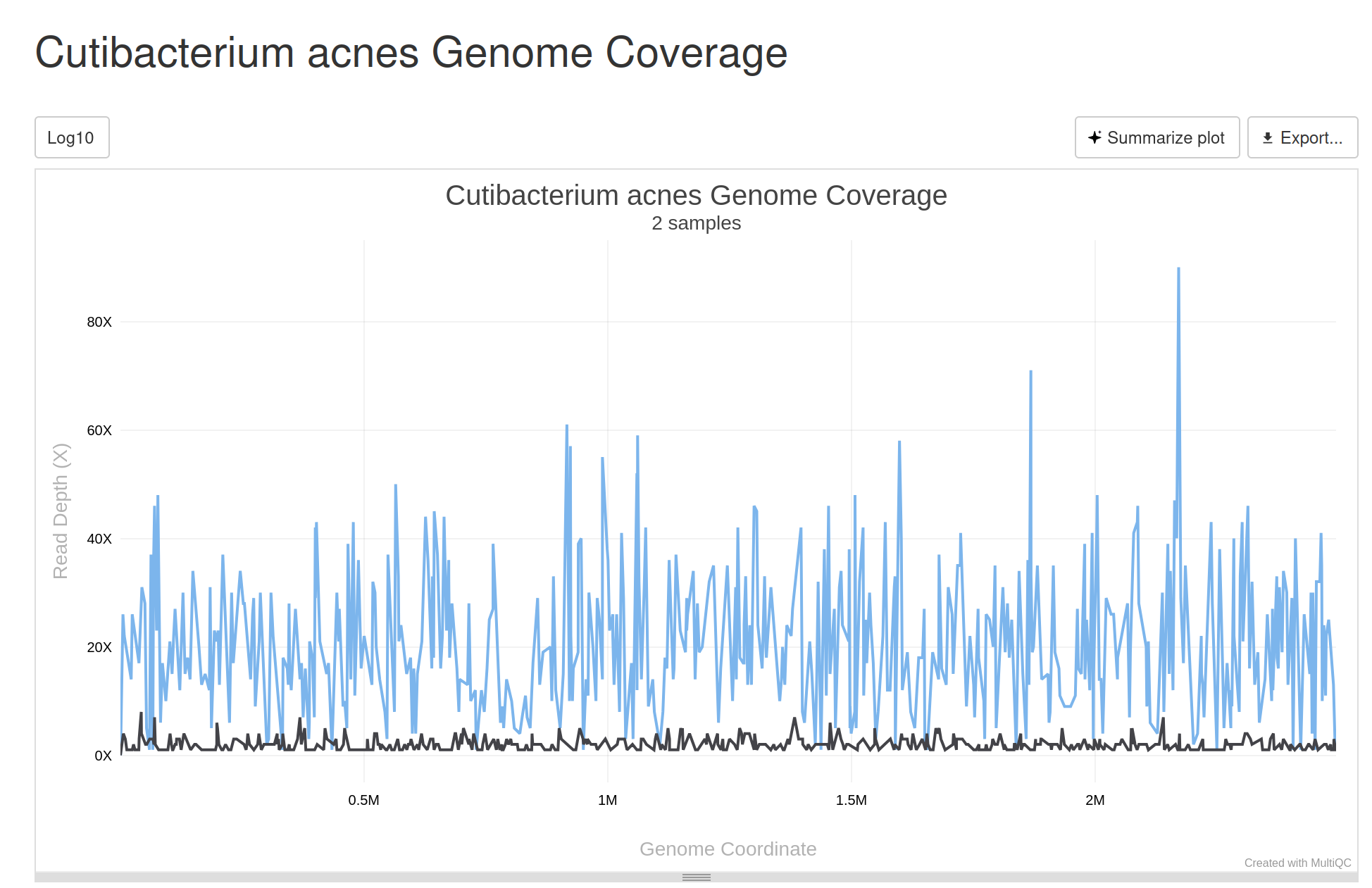
When a pathogen is detected in a complex metagenomic sample, an expert user’s first question is often, “How confident is the detection?” The new genome coverage visualization tool provides a clear, unambiguous answer - providing a clear view of the depth and breadth of sequencing coverage across a pathogen’s reference genome. This plot serves as a transparent quality control metric, allowing a user to assess the confidence of a detection in a complex metagenomic sample. It also enables a user to determine if there was cross contamination of samples by similar genome regions covered across contaminated samples.
High-Resolution Pathogen Typing¶
Differentiating between pathogenic strains and characterizing pathogens using legacy typing schemes (e.g. serotyping) is important for clinical microbiology and public health. The BugSeq platform already provides pathogen-specific analysis for many bacteria, viruses, fungi and parasites (see the Pathogen Pipelines page). Recent updates to the BugSeq platform enhance typing of hypervirulent Klebsiella pneumoniae (hvKP), Influenza A virus, Hepatitis C virus, HIV and Listeria monocytogenes.
Hypervirulent Klebsiella pneumoniae (hvKP)¶

The pipeline now provides more detailed reporting for key K. pneumoniae virulence factors, with a crucial refinement: the platform reports only functional, untruncated proteins as “Detected”.
- By verifying protein integrity, BugSeq provides a result that more accurately predicts the true phenotype, delivering a higher level of clinical actionability.
- The typing of the hypermucoidy-associated rmpADC locus and the rmpA2 gene has been integrated into BugSeq’s core Multilocus Sequence Typing (MLST) engine, ensuring uniform reporting and more robust handling of complex genetic scenarios like truncated or multiple alleles.
Influenza A Virus¶
With the emergence and threat of avian influenza, characterization of Influenza A virus is of paramount importance. Recent updates include:
- Support for typing from single-end Illumina data, broadening compatibility.
- H clade typing, H5 2.3.4.4b genotyping using GenoFLU, and evolutionary origin analysis for H1 and H3 strains with octoFLU.
- These advanced analyses are supported by a significant expansion of influenza genomes in the BugRef database, which improves the sensitivity for all species and serotypes of influenza.
HIV Antiviral Drug Resistance¶
For HIV, the platform now provides variant calling and antiviral drug resistance analysis. Variant calling is performed below the limit of detection of traditional Sanger sequencing (20%), yielding an advantage over Sanger in identifying minority viral populations that can expand under treatment pressure. BugSeq has integrated the Stanford algorithm for a globally accepted prediction algorithm into our secure and private infrastructure. This addition provides critical information that can be integrated into laboratory developed tests.

Listeria monocytogenes¶
For the foodborne pathogen Listeria monocytogenes, BugSeq provides specific typing capabilities to aid in public health and food safety investigations. The platform performs serogrouping for L. monocytogenes, allowing for essential characterization beyond the species level.
Core Accuracy and Database Enhancements¶
Every feature on the BugSeq platform, from taxonomic classification to AMR prediction, is built upon a critical foundation: the BugRef database. The accuracy of any metagenomic analysis is fundamentally limited by the quality of its reference database. While public databases are invaluable resources, they are known to contain sequence contamination, taxonomic misannotations, and other errors that can compromise results. The continuous effort invested in BugRef improvement is what drives the platform’s leading accuracy. We have recently published on some of the key curation methods, and the first half of 2025 saw a series of further updates to BugRef, solidifying this advantage:
- Expanded Fungal and Parasite Genomes: The database was significantly expanded with over 500 new, quality-controlled fungal species that adhere to the MycoBank taxonomy, as well as a large number of curated helminth genomes.
- Improved Bacterial Curation: Strain representation was increased for bacterial species that lack a high number of complete reference genomes, and taxonomic inconsistencies within genera like Providencia were corrected.
- Advanced Quality Control and Refinement: The curation process now incorporates more sophisticated techniques. This includes the implementation of species-specific Average Nucleotide Identity (ANI) thresholds for more accurate species delineation and improved prophage masking. Prophages—viral genomes integrated into bacterial DNA—can confound classifiers, and by computationally identifying and masking them, BugSeq reduces near-neighbor misclassifications. This single improvement was shown in a benchmark to reduce near-neighbor misclassifications by 2-3 per sample.
- Improved Plasmid Detection: The reference plasmid database was curated to remove contaminated sequences and exclude sequences that were likely chromosomal in origin, leading to more accurate detection of plasmids and the AMR genes they often carry.
Advanced Fungal Typing, Genomic Antimicrobial Susceptibility Testing (gAST) and Other Updates¶
H1 saw key advances in other priority areas for our users, including:
- Mycology: ITS genotyping for T. mentagrophytes (T. indotineae identification), and ITS and 28S amplicon reporting from fungal genomes
- AMR prediction (gAST): Large increases in accuracy using a curated database, expert rule curation and mechanistic machine learning
- Short read (e.g. Illumina) 16S/ITS: Expansion of the BugSeq pipeline to support more experimental designs (e.g. multiple amplicons and non-overlapping reads) yielding higher accuracy for the majority of analyses
Given the size and importance of these updates, many will be covered in depth in separate blog posts.
Conclusion: Empowering the Future of Microbiology¶
The H1 2025 updates to the BugSeq platform provide new tools for mycology and bacteriology, improve data interpretation through AI and new visualizations, and include significant enhancements to the core BugRef database. These changes improve the utility of NGS data analysis for microbiology laboratories by increasing the speed, accuracy, and depth of results. They increase diagnostic and surveillance confidence, and provide deeper, more meaningful insights into the pathogens that impact human health every day.
We invite you to explore these features in your BugSeq account today. For those new to the platform, there has never been a better time to see how BugSeq can enable NGS in your microbiology service.