Pipeline change log¶
Latest¶
Added¶
Organism Identification¶
Antimicrobial Resistance¶
- Reporting of topical antimicrobials with EUCAST breakpoints.
Organism Typing & Outbreak Analysis¶
- Option to configure allowable ambiguous loci percentage for outbreak analysis.
- Wastewater SARS-CoV-2 analysis for Ion Torrent data.
Other¶
Fixed¶
Organism Identification¶
Antimicrobial Resistance¶
- Ceftobiprole resistance prediction in the presence of mecA for EUCAST breakpoints.
Organism Typing & Outbreak Analysis¶
Other¶
- Identification of indels for viral consensus sequences in regions with greater than 250X depth.
- Hide internal thinking in some AI summaries.
Changed¶
Organism Identification¶
Antimicrobial Resistance¶
- Hide intrinsic blaOXY genes in Klebsiella genus.
- Better cefepime resistance prediction in the presence of CMY genes
Organism Typing & Outbreak Analysis¶
Other¶
- Improved ONT 16S consensus sequences and sequence identity calculation by factoring in ambiguous bases.
Known issues at release¶
- Mean Phred scores per base in summary report are calculated as a simple average of all base Phred scores, instead of incorporating considerations for a logarithmic scale. Additional details are available in this blog post.
- AMR genes may be reported on a plasmid for drugs which aren’t included in the predicted phenotype panel.
7.0.0 - July 15, 2026¶
Added¶
- Save syndromic panel reports to PDF.
- Median alignment fraction of reads as a configurable metagenomic threshold.
- Several Streptococcus pneumoniae AMR markers including gyrA, parE and rplV variants.
- New user configurable BugID parameter: read alignment fraction against reference genome.
- Application of Corynebacterium breakpoints from CLSI.
- New refMLST schemes for 1667 bacterial species, enabling high resolution outbreak analysis for these species. Previously available schemes remain unchanged.
- New variants conferring AMR in Helicobacter pylori.
- Nextclade typing for RSV, Influenza A H1 segment and Influenza A N1 segment.
- Enable custom pathogen prioritization and report ordering of pathogen calls.
- Relative lineage abundance added for RSV A and B from wastewater samples.
- New drugs reported for M. tuberculosis, including rifapentine and prothionamide.
- Report HIV APOBEC variants and variants of unknown significance.
- AMR prediction models for Staphylococcus epidermidis and Bordetella pertussis.
Fixed¶
- Reported
% Vectormetric in the summary report was missing PhiX reads for short read data. - Chimeric de novo assemblies for ONT ITS data leading to deviations from true taxonomic abundance.
- Detection of AMR genes broken by scaffolding process of de novo assembly. BugSeq assembly bins are now contigs instead of scaffolds.
- Detection of genomic AMR determinants when the drug was reported as intrinsically intermediate.
- Meropenem-vaborbactam resistance prediction in the presence of class B and D beta-lactamases.
- Warning message that RNA viruses detected in DNA sequencing data would be classified as contaminant.
- Influenza type may have been reported as H1N1 if no reads mapped to any H/N segment.
- Detection of AMR genes incorrectly truncated during the gene prediction step.
- Certain gene knockouts would get reported without the
_knockoutsuffix; for example,oprDinstead ofoprD_knockout.
Changed¶
- Improved primer trimming for SARS-CoV-2, HIV and CMV amplicon sequencing data.
- Improved primer masking for SARS-CoV-2 sequences from wastewater sequencing.
- Improved consensus variant calling for all sequencing platforms by implementing an additional variant calling filter for allele fraction.
- Report predicted phenotype for AMR instead of Detected/Not Detected.
- Updating reporting of coronaviruses to common names where possible.
- New BugRef database, bringing:
- Curation of RSV into subtypes, enabling subtyping
- Curation of contaminating 16S, ITS and other amplicon sequences, enabling better classification of reads from these regions
- Labelling of additional viruses to common name instead of binomial species name
- Curation of Betacoronavirus gravedinis
- Inclusion of new fungal and parasite genomes
- New ONT 16S database.
- Improved detection of TruSeq Small RNA adapter sequence.
- Better alignment of reporting of drugs with breakpoints in CLSI M100, including many new drugs reported.
- Use parent folder name for sample naming of Ion Torrent data.
- Update HIV antiviral resistance prediction to Stanford v10.2.
- Improved ONT metagenomic assemblies for data with >97% accuracy, supporting better recovery of multiple strains within a sample.
- Improved circularization of de novo assemblies containing short circular contigs, enabling improved AMR allele identification. For example, BugSeq is now better able to identify blaNDM-1 instead of blaNDM.
- Activate frameshift correction of ONT de novo assemblies in a more targeted fashion, specifically when the number of fragmented BUSCOs exceeds the number of unfragmented BUSCOs.
- Improved ONT 16S accuracy by filtering alignments using query coverage to remove erroneous classifications.
- Improved de novo assembly for Illumina single-end data.
- Update AMR database for M. tuberculosis.
- Improved read preprocessing for MiSeq i100 sequencing data.
- Generation of sample-specific Influenza A reference genome leading to significantly improved consensus sequences.
- Relaxed reference alignment parameters for ONT sequencing data leading to improved viral consensus genomes.
- Updated the drug class label of several drugs for greater consistency.
- Better support for Illumina file naming following Clear Labs’ naming convention.
- Improved AMR prediction accuracy for tobramycin and kanamycin in the presence of aac(3)-II member.
- Improved detection, splitting and reporting of mixed bacterial and fungal isolates by leveraging additional features to determine if the isolate is pure or mixed.
- AMR database update.
- Update to mechanism for detecting AMR gene knockouts, which brings increased accuracy.
- Stop reporting Mycobacterium avium, Mycobacterium intracellulare and Treponema pallidum subspecies until further availability or curation of type strains is available.
- Improved cephamycin resistance prediction in the presence of ESBLs.
- Polish ONT assemblies. In internal benchmarks, we observed a ~50% reduction in assembly errors due to basecalling errors from methylation.
- Improved curation of Enterobacter hormaechei subspecies.
- Selectively report antimicrobials for potential agents of bioterrorism.
- Upgrade AI model for improved report summaries.
- Ensure consistency for MLST reporting: only report schemes with at least one locus detected.
- Assemblies with a high fraction (default: 10%) of ambiguous loci are excluded from refMLST (high-resolution) outbreak analysis.
Known issues at release¶
- Mean Phred scores per base in summary report are calculated as a simple average of all base Phred scores, instead of incorporating considerations for a logarithmic scale. Additional details are available in this blog post.
- AMR genes may be reported on a plasmid for drugs which aren’t included in the predicted phenotype panel.
6.1 - Feb 5, 2026¶
Added¶
- Many new amplicons for in silico PCR from de novo assemblies, including D1D2, Calmodulin, Elongation Factor 1α, β-tubulin, and IGS for Fungi, as well as 16S, HSP65 and rpoB for novel bacterial species.
- Several promoter mutations conferring AMR in P. aeruginosa.
- Support for PacBio 16S sequencing data.
- Modeling of susceptible dose dependent phenotype for AMR prediction when using CLSI predictions.
Fixed¶
- Colors in reports when in dark mode.
- Rare scenario where viral contigs were classified to species name (for example, Lentivirus humimdef1) instead of the common name in NCBI (for example, Human immunodeficiency virus 1)
Changed¶
- Remove ONT demultiplexing in BugSeq based on barcodes in read sequences. The recommended workflow is to demultiplex before submission using MinKNOW/Dorado. BugSeq continues to extract barcode and barcode alias information from ONT filenames and read headers.
- Update reporting framework (MultiQC) to enable dark mode in reports.
- Update drug class naming to conform with CLSI M100 drug classes.
- Implemented multiple iterative rounds of adapter and barcode trimming for ONT datasets to better remove repeated adapters and barcodes at read ends. A side effect of this change is the preprocessing statistics in the Summary Report no longer reflect the reasons for read filtering (for example, too long, too short, low complexity, etc.).
- Update NCBI core_nt database.
- Updated BugRef database, restoring certain viral species such as HIV-1 which were inadvertently removed from the database in v5.8.
- Improved Genome Coverage visualization by joining all reference contigs into a single visualization.
Known issues at release¶
- Mean Phred scores per base in summary report are calculated as a simple average of all base Phred scores, instead of incorporating considerations for a logarithmic scale. Additional details are available in this blog post.
- AMR genes may be reported on a plasmid for drugs which aren’t included in the predicted phenotype panel.
6.0 - Jan 6, 2026¶
Added¶
- Report ceftolozane-tazobactam prediction for Pseudomonas aeruginosa AMR prediction.
- Many new organisms as potential pathogens in CSF samples.
- Report median alignment length of reads against the reference genome for each detected taxon.
- Candida parapsilosis MLST.
- Detection of MabTetX for tetracycline resistance in M. abscessus.
- Report minocycline AMR prediction for M. abscessus.
- Ability to select EUCAST for AMR phenotype predictions. Predictions follow EUCAST v15.0 breakpoints and are intended to match the latest EUCAST breakpoints. The default BugSeq predictions follow the latest CLSI standards (for example, M100, M45, etc.) - this remains unchanged. Get in touch with us to enable early-access EUCAST predictions for your account.
- Report intrinsic resistance and intermediate AMR for a broad range of organisms.
- Ability to export Newick format from outbreak visualization to visualize outbreaks in external tools.
- blaZ typing.
- Detection of a mutation in blaZ(A) conferring cefazolin inoculum effect for Staphylococcus aureus.
- Many variants conferring intermediate vancomycin phenotype in Staphylococcus aureus.
- Adapter prevalence plot for ONT data in the Summary Report. High adapter rates indicate that adapter/barcode trimming may not be enabled in MinKNOW. It’s highly recommended to enable this setting. Future versions of BugSeq will remove all ONT adapter/barcode trimming functionality.
- New reported drugs for P. aeruginosa and Acinetobacter.
Fixed¶
- Insertion sequence detection for Acinetobacter baumannii AMR prediction occasionally detecting insertion sequences not directly upstream of the AMR gene.
- Detection and basecaller errors for R9.4.1 PromethION samples with super accuracy basecalling.
- Detection of AMR genes found in circular bubbles of the assembly graph. This change applies to short read metagenomic sequencing only.
- Depth of Sequencing columns in General Statistics (≥20X and ≥40X) would occasionally show 0% when the depth was higher than this threshold. See this issue and fix from the BugSeq team on GitHub for details.
- Reporting of appropriate antifungals for C. auris AMR prediction.
Changed¶
- Major improvements to pathogenicity prediction algorithm: Pathogenicity reporting is now based on the interaction of organism, sample type and detection thresholds. CSF, upper respiratory and lower respiratory sample types leverage machine-learning classifiers for thresholds. All other sample types use a default 1% abundance threshold. Custom threshold application has also been released and can be configured in the “Lab” settings. Read a recent blog post on the innovation underlying this change.
- Apply multiple MLST schemes for M. abscessus.
- Downgrade pathogenicity prediction of cellular and acellular root-ranked taxa.
-
Updated BugRef database, bringing:
- More accurate and more frequent identification to the subspecies rank for select species.
- Greater species representation of fungi by leveraging additional sources of information to confirm the identity of public genomes.
- Manually curated Cunninghamella genomes.
-
Improved prediction of cefiderocol resistance in K. pneumoniae, E. coli and E. cloacae with the addition of several new curated rules.
- Improved AMR prediction for all members of the S. aureus and N. glabratus (formerly known as C. glabrata) complexes.
- Don’t automatically render plots on load of Genome Coverage visualization to handle reports with thousands of plots.
- Upgrade AI model for improved report summaries.
- Remove warning of the presence of RNA viruses for DNA sequencing of bacterial/fungal isolates. RNA viruses may still be reported in the Krona plot and metagenomic classification files, but there will no longer be a warning at the top of reports.
- Report the insertion sequence type and gene allele when they predict resistance in A. baumannii.
- Improved host range prediction of plasmid cluster AD094.
- Updated AMR database bringing hundreds of new gene alleles and several new knockout genes. Correlation with phenotype on internal benchmarks is improved across all drugs.
- Improved curation of mutations causing resistance for P. aeruginosa.
- Improved prediction of carbapenem resistance in A. baumannii by detecting insertion sequences upstream of intrinsic OXA beta-lactamases.
- Improved isolate identification by decreasing the relative fraction of the assembly required to call the identity to 50%, which matches the updated Genome Taxonomy Database threshold.
- Improved identification of M. intracellulare and M. paraintracellulare by setting a custom ANI threshold.
- Filter genomes failing QC thresholds before outbreak analysis. This change ensures that allele differences in outbreak analysis aren’t the result of lower quality input data.
Tip
If genomes aren’t included in outbreak analysis, review the per-sample report to determine which quality assessments failed. Quality control thresholds may be adjusted in Lab settings. BugSeq has set defaults which are applicable to many sequencers, organisms, and labs. These standards also largely conform to standards such as CDC’s PHoeNIx.
- Drop S. epidermidis virulence factor reporting.
- Calculate isolate quality control statistics only on contigs greater than 500bp, similar to CDC’s PHoeNIx. This change results in a greater number of samples passing quality control.
- Improved accuracy of completeness assessment for members of the family Yersiniaceae by using a family-specific model.
- Improved carbapenem resistance prediction for E. cloacae by searching for porin knockouts.
- Improved macrolide resistance prediction across all organisms.
- Use barcode aliases as barcode names for ONT sequencing data if they were used during the sequencing process.
- MLST scheme database update.
- Improve detection of MabTetX and erm(41) AMR genes when they’re less than 98% similar to the reference sequence.
- Update pangolin and pangolin database.
- Improved de novo assembly and reference-based depth calculation. Previously, BugSeq relied on mosdepth, which ignores reference sequences with no reads aligned (see GitHub issue #87, #180 and #198 and #222 for details of the issue). We’ve now implemented custom logic which is more accurate. This change impacts any table or plot with depth calculations, including summary reports, per-sample reports and genome depth visualization.
- Updated and expanded reference genome database used for reference sequence alignments and depth calculation. This database now included representation of fungi and parasites not found in RefSeq.
- Update the minimum suggested basecaller version for R10.4.1 ONT sequencing to v5.2.0.
- Closer alignment of AMR drug reporting to drugs with available breakpoints. The largest change impacts Streptococcus pneumoniae with reporting of meningitis/non-meningitis breakpoints.
- Distribute plasmid contigs to all taxonomic children of the predicted host range of the plasmid. Previously, these contigs were left at higher taxonomic ranks if there were multiple possible host bacteria.
- Update reference plasmid database. Plasmids with host range Staphylococcus have been curated.
- Contigs may be placed in multiple taxonomic bins. This change improves binning of sequences with high sequence homology between multiple organisms in the sample. Reads may therefore also be placed in multiple taxonomic bins. Fractional reads are used to calculate abundance and rounded to the nearest whole read before reporting.
- Add several common genuses as contaminants to pathogenicity algorithm.
- Change to how negative control multiplicity is calculated: the denominator in the sample and negative control is now
total reads - host reads. This change enables better control for varying fractions of human reads in a sample. The negative control multiplicity is therefore now the abundance of a taxon relative to other microbes. - Improved adapter and barcode trimming for ONT data by automatically detecting overrepresented sequences at read ends. BugSeq continues to recommend that demultiplexing and trimming be performed before submission for optimal results.
- Better automatic detection of ONT 16S data by preprocessing reads before detecting experimental design.
Known issues at release¶
- Mean Phred scores per base in summary report are calculated as a simple average of all base Phred scores, instead of incorporating considerations for a logarithmic scale. Additional details are available in this blog post.
- AMR genes may be reported on a plasmid for drugs which aren’t included in the predicted phenotype panel.
5.9 - Feb 3, 2026¶
Added¶
Fixed¶
Changed¶
- Reversion of BugRef curated reference database to v5.7 database to address missing viral species such as HIV-1.
Known issues at release¶
- Mean Phred scores per base in summary report are calculated as a simple average of all base Phred scores, instead of incorporating considerations for a logarithmic scale. Additional details are available in this blog post.
- AMR genes may be reported on a plasmid for drugs which aren’t included in the predicted phenotype panel.
5.8 - Sep 22, 2025¶
Added¶
- ITS genotyping for Trichophyton mentagrophytes.
- Report depth requirement in QC column header for each variant in HIV resistance prediction.
- Doubled the number of plasmids in BugRef for improved plasmid classification.
- New AMR models:
- Metamycoplasma hominis (formerly known as Mycoplasma hominis)
- Mycobacteroides abscessus (formerly known as Mycobacterium abscessus)
- T2 phage as an internal control which is automatically detected.
- Report mean read length in General Statistics table of summary report.
- Report the number of unclassified reads from nanopore 16S samples.
- Added three new blaSHV alleles to AMR database.
- Warnings to summary reports for low quality ONT input data, such as from outdated basecaller models, and
fastandhacbasecalled data. See the Basecalling docs for maximizing input data quality. - Nextclade results to per-sample reports.
- Add sulbactam/durlobactam reporting for A. baumannii.
Fixed¶
- HIV resistance variants found at high relative abundance but not low relative abundance.
- Depth of sequencing used for Illumina consensus variant calling. See the BCFtools GitHub issue from our team for details.
- HIV variants with multiple potential variants at the same site getting called as only one of the potential variants. See the GitHub issue for corresponding details.
- Update reference genome database, fixing missing reference genome alignments and unique read alignments.
- SARS-CoV-2 lineages all getting reported as lineage
A.
Changed¶
- Update taxonomy database.
- Update BugRef, bringing:
- Improved curation of species with species-specific ANI thresholds.
- Improved handling of prophage sequences reducing near-neighbor misclassifications. In a benchmark of monomicrobial metagenomic samples, we saw a reduction of 2-3 near-neighbor misclassifications per sample.
- Update 16S database based upon above BugRef update.
- Update AI model for improved AI summaries.
- Change reference genome for HIV-1 from NC_001802.1 to K03455.1 to harmonize with external services such as Los Alamos National Laboratory.
- Improved prediction of AMR from protein knockouts in incomplete genomes.
- Improved classification of viruses to common names instead of scientific species names.
- Improved genome completeness assessment for Helicobacter genomes by using specialized BUSCO dataset.
- Lowest abundance for HIV resistance set to 10% minority variants. Contact support if your lab needs a different threshold.
- Suppress more intrinsic AMR genes for many bacterial taxa including P. aeruginosa, Klebsiella species and ampC producers, resulting in improved phenotype prediction.
- Improved generalizability of machine learning-based AMR rules to cases not seen in the BugSeq training database.
- Improved application of machine learning-based AMR rules by using species-specific models instead of organism group models.
- Report colistin as intrinsically intermediate for certain organisms according to CLSI M100.
- Enabled read deduplication for C. auris analysis (note this doesn’t impact MycoSNP results).
- Improved handling of variants in primer regions for viral variant calling and consensus sequence generation.
- Improved genome completeness assessment of taxa within Chlamydiota.
- Enable reporting of short-read 16S to species group rank.
- Improved automatic detection of short-read 16S experiments for application of the optimal analysis parameters.
- Increases in pathogenicity prediction for Staphylococcus aureus (all sample types), and Adenovirus (CSF).
- Report more viruses using their common name instead of species name. For example, report “Eastern equine encephalitis virus” instead of Alphavirus eastern.
- Integrate IUPAC ambiguous bases into short read assemblies to mitigate impact of collapsed repeats.
- Improved classification of bacterial isolates to the subspecies rank.
- Improved phenotype prediction for ambiguous AMR gene alleles of blaSHV, blaTEM and blaGES.
- Updated HIV-1 antiviral resistance prediction database.
- Downgrade pathogenicity prediction of high ranks such as root, domain, kingdom, phylum, class and order.
Known issues at release¶
- Mean Phred scores per base in summary report are calculated as a simple average of all base Phred scores, instead of incorporating considerations for a logarithmic scale. Additional details are available in this blog post.
- AMR genes may be reported on a plasmid for drugs which aren’t included in the predicted phenotype panel.
5.7 - May 20, 2025¶
Added¶
- IQ-TREE phylogenetic tree outputs for MycoSNP.
- Extract and report ITS and 28S rRNA amplicons from fungal assemblies.
- Influenza typing for single-end Illumina data.
- A significant number of high quality, curated helminth genomes to BugRef reference sequence database.
- Increased strain representation in BugRef for bacterial species without complete genomes, further improving classification performance.
- More vector sequences to BugRef found to contaminate some of our users’ data.
- Virulence factor reporting for Streptococcus pneumoniae, Streptococcus pyogenes (Group A Strep) and Streptococcus agalactiae (Group B Strep).
- Suggested taxon to report for ONT 16S based on identity of the consensus sequence against the reference sequence.
- Reporting of the number of nonoverlapping genomic intervals with read coverage for short read data in the per-sample report General Statistics table. This addition compliments the existing unique read alignment column, which contains overlapping and non-overlapping alignments with different start coordinates.
- Average nucleotide identity reporting for de novo assemblies in the per-sample reports, under “Assembly Statistics” section.
- Influenza A virus H clade typing.
- Influenza A virus H5 2.3.4.4b genotyping with GenoFLU.
- Influenza A virus H1 and H3 evolutionary origin analysis with octoFLU.
- AI interpretation in interactive reports.
Note
AI interpretations should be correlated with the content of reports for confirmation of interpretation. AI interpretations can be turned off in your BugSeq Lab’s settings.
- Genome coverage visualization for metagenomic samples. This output is located under the “Other” dropdown. Note that large numbers of taxa detected across many samples may require a powerful computer to view.
Example Genome Coverage Visualizer
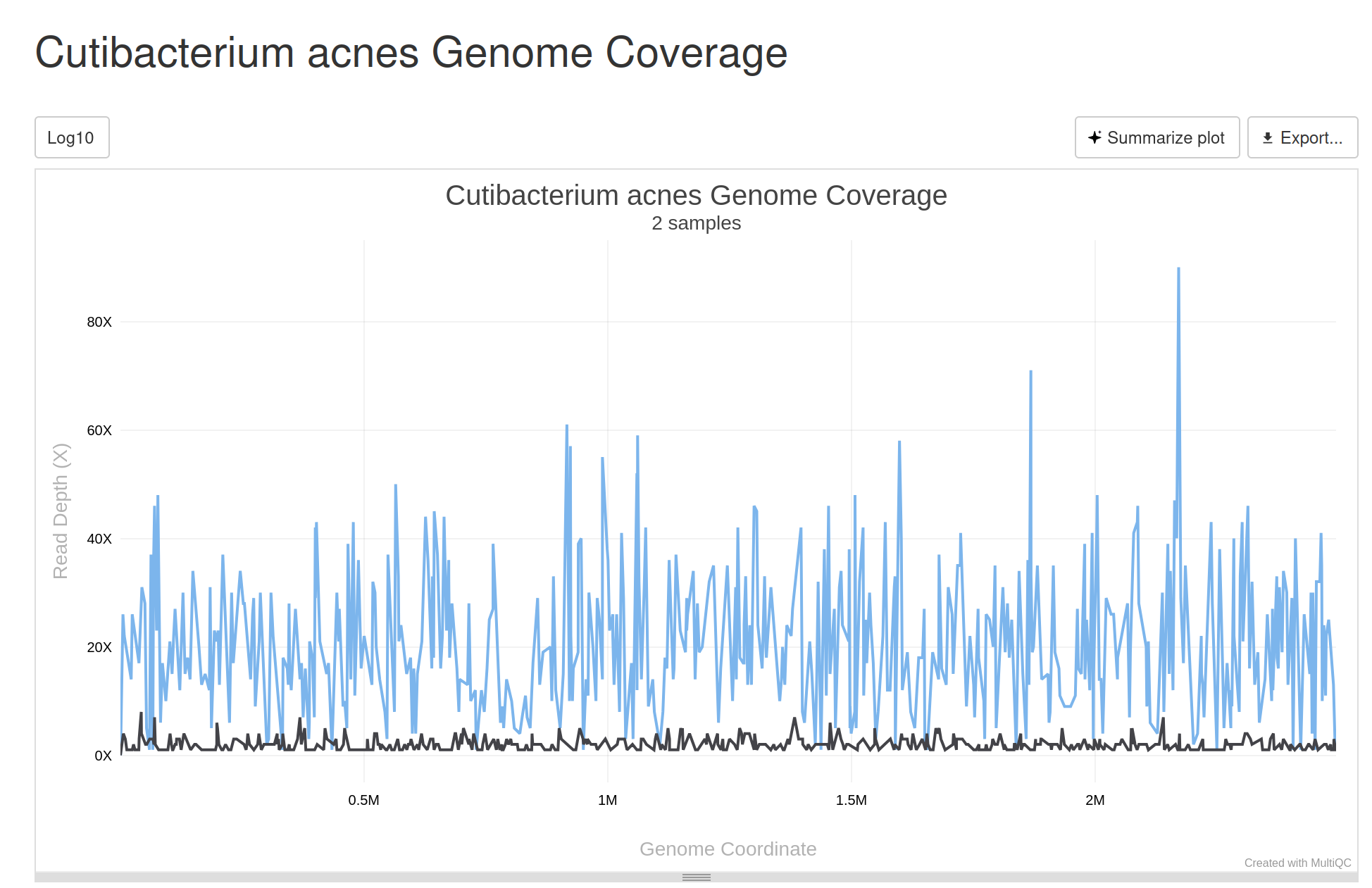
- Listeria serogrouping.
- Report canonical SNP typing results in summary report.
- Report host and vector filtering statistics from Nanopore 16S.
- Hepatitis C virus genotyping from short read sequencing data.
- Increase the number of genotypic determinants for P. aeruginosa AMR prediction. Twenty protein knockouts and 156 amino acid variants have been added. Categorical agreement is improved with a decrease in very major error rate.
- Outbreak analysis now has an option to show sample labels (and metadata labels) on the visualization.
- Overlay metadata on outbreak analysis to visualize outbreaks across facilities, wards and more.
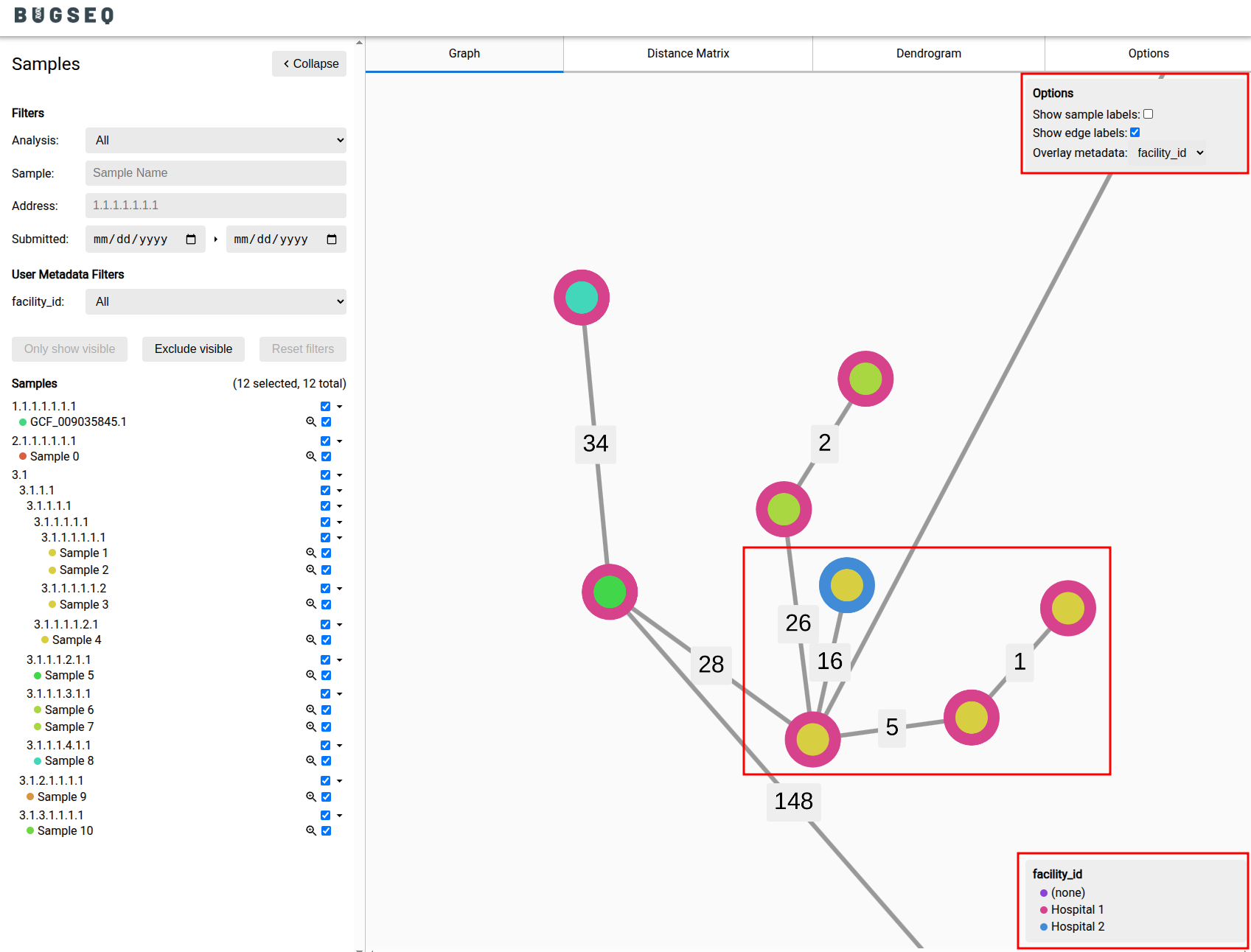
- Results emails and per-sample reports now report outbreak cluster size (if cluster alerting is configured).
Fixed¶
- Bug inflating unique read alignment count. See the fix by BugSeq on GitHub for details.
- Prohibit special characters in sample metadata for better outbreak analysis stability, and properly handle special characters that have already been submitted to BugSeq and saved.
Changed¶
- Improved AMR model such that reported genomic predictors are more accurately associated with phenotype.
- Improved adapter detection and trimming for Illumina single-end samples with very low rates of adapter contamination (<1% of reads).
- Improved adherence to CLSI M24S guidelines for reported drugs for Mycobacteria and Nocardia.
- Improved cephalosporin resistance for E. cloacae complex based on ampD knockout.
- MycoSNP now runs using a clade-specific reference genome. This change reduces the number of erroneous SNPs seen for isolates from clades II-VI. Previously, a clade I reference genome was used for variant calling as this is the reference included with MycoSNP. All historical lab data will now be included in the clade I outputs, while new data will be found in clade-specific outputs.
- Improved identification and masking of prophage sequences in BugRef by combining multiple detection approaches.
- Improved quality control of Streptococcus pneumoniae serotyping results. The quality control flag and comment now incorporate the confidence of the prediction beyond the assembly quality.
- Improved selection of samples to visualize upon initial loading of the outbreak visualizer. The current analysis’ samples, along with those within 50 alleles, are now displayed by default.
- Deterministic loading of the minimum spanning tree in the outbreak visualizer.
- Isolate summary output is renamed from
bacterial_isolate_summary.xlsxtoisolate_summary.xlsxto reflect that it also includes fungal isolates. - Include organism name along with cluster name in report headers.
- Expanded AMR database with additional markers of resistance for nitrofurantoin in E. faecium.
- Improved short read 16S, which makes fewer assumptions about user data. Paired-end reads which don’t merge and off-target amplification are now better supported and classified. The improved 16S pipeline also brings per-sample reports to harmonize reporting with other experimental designs.
- Improved automatic detection of combined 16S/ITS Illumina samples.
- Improved AMR prediction for Mycoplasmoides pneumoniae.
- Updated AMR database, bringing >400 new genes and alleles.
- Update BUSCO for improved completeness/contamination checks using
odb_12datasets. - Update Pangolin database.
- Improved detection of C. auris clade for MycoSNP analysis.
- Improved short read 16S profiling performance so that fewer low abundance false positive taxa are reported.
Known issues at release¶
- Mean Phred scores per base in summary report are calculated as a simple average of all base Phred scores, instead of incorporating considerations for a logarithmic scale. Additional details are available in this blog post.
- AMR genes may be reported on a plasmid for drugs which aren’t included in the predicted phenotype panel.
5.6 - Feb 10, 2025¶
Added¶
- Report read coverage used in metagenomic classification. These results are found in
Metagenomic Classification>Read-Based>DetailsCSV files. - E. coli pathotyping.
- Reporting of Klebsiella pneumoniae virulence factors. See a recent report from Russo, Lebreton & McGann for details on the reported virulence factors.
Note
Only functional (untruncated) proteins are reported as “Detected”. As per Lam et al. (2024), “the rmpA and rmpA2 genes appear to be frequently subjected to insertions or deletions (indels) within a poly(G) tract that consequently encode a truncated and presumably non-functional product…Importantly, these loss of function mutations also need to be carefully considered when interpreting data based solely on PCR detection of rmpA/A2, and may partly explain the discrepancies in the literature reporting on the association between rmp presence and [hypermucoviscosity].”
- Dendrogram visualization for outbreak analysis.
- Date filtering for outbreak analysis visualization. Note that default filters are now applied to the visualization to include samples from the most recent six months, however these can be adjusted to retain previous functionality of viewing all historical data.
- Report virulence factors for Staphylococcus epidermidis.
- Added a significant number of influenza genomes to BugRef database to improve sensitivity for all species and serotypes of influenza.
- Module to handle low complexity tails of ONT reads, resulting in improved metagenomic accuracy for ONT data.
Fixed¶
- Detection of multiple alleles of E. coli shiga toxin present in a sample sequenced with a long-read sequencer.
- Detection of very fragmented AMR genes in assembly where genes are fragmented into parts comprising less than 50% of the reference sequence.
Changed¶
- Better detection of divergent viruses from the reference database sequences by separately considering sequence divergence and uniqueness of read classification. Previously these metrics were combined into a single metric used internally in classification.
- Report host-filtered reads as Homo sapiens instead of Chordata if “Filter animal reads” wasn’t selected upon data submission.
- Sequence typing of Klebsiella pneumoniae rmpADC locus and rmpA2 has moved to the Multilocus Sequence Typing section and no longer relies on Kleborate to perform typing. The change from Kleborate to BugSeq’s MLST approach enables uniform reporting with all other MLST schemes (relevant docs), as well as more accurate results in the case of truncated, missing or multiple alleles found in a sample. rmpADC locus and rmpA2 loci information is no longer reported in the Klebsiella pneumoniae typing section.
- Update MLST database.
- Improved descriptions in reports for Klebsiella pneumoniae typing, antimicrobial resistance and virulence factor detection.
-
New BugRef database, incorporating:
- Addition of >500 new fungal species which have undergone rigorous quality control.
- Adherence to accepted fungal taxonomy of MycoBank, one of the three global fungal name registration repositories recognized by the Nomenclature Committee for Fungi and ratified by the International Mycological Association.
Notable changes to BugSeq’s results
- Trichophyton indotineae is now classified as Trichophyton mentagrophytes
- Candida auris continues to be named Candidozyma auris since BugSeq v5.5
See MycoBank simple search for details.
- Improved quality control of all genomes for contamination.
- Correction of taxonomy within Providencia genus for identical species with different names.
-
New 16S databases based on the above BugRef database update. Additionally, improved curation of Bacillus cereus group 16S sequences for improved full-length 16S classification accuracy.
- New GC and length distribution calculation for each species, reducing errors with quality control thresholds. NCBI rounds their values which was causing imprecision and therefore inappropriate flags in genome quality control. By generating these statistics, BugSeq overcomes the limitations of using NCBI to get expected GC and length distributions for each species.
- Improved 16S performance for ONT R10.4.1 reads basecalled with SUP model.
- Updated reported antimicrobials to conform with CLSI M100 ED35:2025.
- Unique read alignments for Illumina sequencing now uses both nucleotide and amino acid alignment for increased alignment sensitivity. This upgrade improves results for samples with pathogens which have divergent genomes to the reference genome.
- Improved precision for detection of Mycobacterium tuberculosis complex from metagenomic data by using a custom nucleotide identity threshold.
- Add zoonoses like Capnocytophaga canimorsus and Pasteurella multocida to the predicted pathogens list.
- Improved prediction of cefiderocol resistance in E. coli.
- Upgraded pathogenicity prediction of C. auris for all sample types.
- Improved detection of adapters for Illumina single-end data where the adapters were present in less than 1% of reads. This change improves precision of metagenomic classification.
Known issues at release¶
- Mean Phred scores per base in summary report are calculated as a simple average of all base Phred scores, instead of incorporating considerations for a logarithmic scale. Additional details are available in this blog post.
- AMR genes may be reported on a plasmid for drugs which aren’t included in the predicted phenotype panel.
5.5 - Dec 15, 2024¶
Added¶
- Generate genome alignments, coverage information and unique read counts for protozoa.
- Generate per-sample reports for nanopore 16S samples.
- MLST for Serratia, Proteus and Providencia species.
- Report genomic predictors of resistance for mupirocin in Stapylococcus aureus.
- Report taxonomic lineage of organisms in General Statistics table of per-sample reports. This column is hidden by default.
- Genome assembly quality control for Trichophyton and related fungi.
- Run both antimicrobial resistance and SARS-CoV-2 mixture analyses on wastewater.
- Norovirus genotyping.
- Additional QC flags for bacterial isolates, including deviation from species distribution for assembly length and GC content.
- MLST analysis for fungi including C. albicans, C. krusei, C. glabrata and others.
- Cryptosporidium GP60 subtyping.
- Report hypermucoidy locus information for Klebsiella pneumoniae.
- Report additional details and outputs from ONT 16S analyses, including per-read taxonomic classifications and a joined taxonomic classification table including all samples.
- HIV antiviral drug resistance prediction.
- Execute both published MLST schemes for V. cholerae.
- Add cerebrospinal fluid (CSF) option on data submission for tailored CSF pathogen identification.
- Virulence factor reporting for many species including Stapylococcus aureus, Vibrio cholerae, Corynebacterium diphtheriae, Clostridioides difficile and more.
Fixed¶
- Genomes with multiple MLST alleles for a locus, for example, 1 and 200, were reported as
locus_a(1 200). This reporting structure could have been confused with allele1200. A/has now been added between multiple alleles and it’s reported aslocus_a(1/200). - Correct detection of viruses with RNA genomes. Previously, some viruses with DNA genomes, like Hepatitis B virus, may have been reported with an RNA genome.
- Drugs reported for Campylobacter jejuni and Campylobacter coli now follow CLSI M100 guidance.
- Fix combination knockout like nfsA/B, receiving high confidence AMR prediction from incomplete genomes. Confidence for knockouts is now based on the completeness of the genome.
- E. coli serotyping from paired-end short read data was reported as a failure to identify serotype.
Changed¶
- Pin first column in report tables.
- Improved performance for detection of genotypic predictors of trimethoprim-suflamethoxazole resistance prediction in Acinetobacter baumannii.
- Hide many intrinsic AMR genes from being reported. For example, the intrinsic
aac(6')is no longer reported Serratia species. Non-intrinsic alleles and genes will continue to be reported. - Improved de novo assembly quality for short reads by removing artifactual contigs after assembly.
- Updated MLST database.
Warning
While we’ve overall seen more organisms with sequence types called with the updated database, we’ve also observed several cases where sequences types are no longer able to be called. This situation arises when new alleles in the MLST database cause multi-allele detection where a locus was previously detected as a single allele. The loci alleles can be inspected to identify the locus with the new multi-allele. We’ve also observed other cases of alleles being removed from the MLST database causing failure to produce a sequence type with the updated database. BugSeq follows the accepted PubMLST schemes and issues with these schemes should be directed to their maintainers as listed on PubMLST.
- Improved taxonomic classification of contigs with very low average nucleotide identity by requiring a minimum proportion of bases to uniquely match.
- Updated reference plasmid database to remove a contaminated sequence (plasmid cluster AE985).
- For bacterial isolate analyses, in the General Statistics table of the summary report, report isolate taxon and percentage of reads assigned to isolate taxon instead of the top organism across all samples and percentage of reads assigned to top 10 species.
- Warn when GC content of bacterial isolates doesn’t follow a normal distribution.
- Improved taxonomic classification of contigs by implementing a two step algorithm, first profiling assembled organisms and then binning to high confidence detections.
- Improved taxonomic classification of short read data by implementing logic to leverage the taxonomy, assembly graph and contig coverage for contig classification.
- Downgrade pathogenicity of E. coli from sterile samples to neutral. Future work will bring a more nuanced prediction of pathogenicity for E. coli.
- Improved warnings based on GC content for bacterial and fungal isolates.
- 16S analyses now use a 16S database curated by the BugSeq team. This database provides large advantages over alternative databases like SILVA, NCBI and others including:
- Species-level curation
- Adherence to NCBI Taxonomy
- Improved representation of intra-species sequence variation
- Removal of contaminated and erroneous 16S sequences
Accuracy
These advances bring more accurate 16S analysis across all sequencing platforms.
- New 16S analysis for ONT. This analysis brings:
- Improved species-level resolution
- Integration with the BugSeq-curated 16S database
- Support for reads shorter than the full 16S locus
- Generation of a single representative sequence per species, enabling comparison of sequence identity and coverage to reference sequences which is reported in per-sample reports
- Update Pangolin database for SARS-CoV-2 lineage typing.
- Improved Nanopore metatranscriptomic (RNA) taxonomic classification by filtering poor alignments of reads back to the de novo assembly.
- Curation of relaxase sequences in plasmid databases by removing several erroneous transposase sequences, improving plasmid detection and mobility prediction.
- Improved bacterial isolate assembly across all platforms by better handling low complexity reads in preprocessing steps.
- Improved bacterial isolate assembly from short read data by leveraging an additional assembler in the BugSeq pipeline and tuning it for maximal accuracy.
- Upgrade pathogenicity prediction of Cronobacter species from CSF samples.
- Additional steps for identification of low quality genomes in BugRef.
- Increased strain representation for many species in BugRef.
Details
BugSeq has benchmarked this change across >300 isolates with high quality reference genomes available. Overall, it reduces the error rate of our assemblies by a median of 2 SNPs per 100kb, 1 indel per 100kb and 3 misassemblies per genome. BugSeq assemblies now achieve near maximal accuracy possible from short read data. Users can expect to see a minor reduction in allele distances in outbreak analysis (refMLST), as most of these errors were previously clustered within very few genes. Not all species were impacted equally, with species containing large numbers of repeats seeing greater quality improvements. Plasmid identification and recovery from short read assemblies performed similarly with the new approach and shouldn’t be impacted.
- Better AMR prediction for novel and inexact beta-lactamase matches.
- Offer NCBI core_nt database instead of nt as an alternative database option. This change is consistent with recommendations from NCBI and changes in the default behavior of BLAST, and yields faster BugSeq analysis compared with NCBI nt. Read more about the change in the NCBI blog post. For almost all applications, we continue to recommend our default BugRef curated database, which yields more accurate and faster results.
- Curate genomes in BugRef with contaminating 16S rRNA sequences. This changes improves classification performance of reads deriving from the 16S rRNA locus.
- Update NCBI Taxonomy database. Note that Candida auris has been renamed to Candidozyma auris (NCBI Taxonomy page, supporting citation).
- Split reporting of C. difficile toxins into A and B instead of combining them together as a single virulence factor.
Known issues at release¶
- Mean Phred scores per base in summary report are calculated as a simple average of all base Phred scores, instead of incorporating considerations for a logarithmic scale. Additional details are available in this blog post.
- AMR genes may be reported on a plasmid for drugs which aren’t included in the predicted phenotype panel.
- E. coli shiga toxin detection may miss multiple alleles if both occur in the same sample and data was generated with a Nanopore or PacBio sequencer.
5.4 - Sep 13, 2024¶
Added¶
- Reporting of text warnings at the top of reports for anomalous data. The first warning implemented is triggered for RNA viruses detected in DNA samples.
- Reporting of internal control reads per million in the General Statistics table.
- Reporting of normalized read counts relative to internal and negative controls.
- Reporting of assembly completeness in the summary report for bacterial isolates.
- Support for Illumina single-end reads.
- Depth reporting for bacterial isolates with Illumina single-end.
- Run assembly completeness/contamination checks on more organisms, including some Herpes viruses.
Fixed¶
- Assembly completeness coloring is fixed to more logical colors (see MultiQC Issue #2450).
- Running the same sample through BugSeq multiple times on the same analysis version rarely resulted in different plasmids detected. The issue was reported by BugSeq and a fix implemented in collaboration with the upstream tool maintainers.
- Remove the hidden average and median read length columns in the General Statistics table of summary reports. Users previously could enable visualization of these columns, which would show inaccurate data; inaccuracy was the result of how MultiQC calculated them.
- Selection of MLST schemes for Klebsiella aerogenes and Enterobacter cloacae complex members. Rarely, a Cronobacter MLST scheme would be automatically selected instead of the appropriate MLST scheme.
- Use lineage-specific BUSCO datasets where a more general dataset doesn’t produce accurate results.
Changed¶
- AMR database update.
- Improved AMR prediction for fosfomycin in Klebsiella pneumoniae by selective reporting of fosA alleles.
- Improved aminoglycoside AMR prediction.
- Report 10 instead of 5 top taxa in the metagenomic classification plots in the summary report
- Better protein knockout detection for AMR prediction.
- Read classification CSV outputs classification of host range of plasmid, regardless if the sample is a bacterial isolate or not. This change ensures the read classification CSV matches results from the krona plot and reports.
- Improved reporting of viruses by using their common names when appropriate. For example, Influenza A virus will be used in many places instead of Alphainfluenzavirus influenzae. See the NCBI Insights post for details.
- Improved annotation of blaSHV alleles from Tsang et al.
- Improved prediction of nitrofurantoin resistance in E. coli based on Dulyayangkul et al.
- Improved prediction of carbapenem resistance in Pseudomonas aeruginosa by curating public mutations.
- Improved prediction of vancomycin resistance in Enterococcus species by requiring select combinations of van operon genes. See Coll et al. for details.
- Improved Nanopore host filter by using dynamic filtering criteria based on alignment length.
- Improved prediction of beta-lactam resistance in Acinetobacter calcoaceticus/baumannii complex by incorporating information on intrinsic beta-lactamases and insertion sequence detection into prediction.
- BugRef (curated reference sequence) database updated. Fungal representation is improved including manually curated Trichophyton genomes from the T. mentagrophytes complex.
- Improved which drugs are reported for agents of bioterrorism to match CLSI M45-A3.
- SARS-CoV-2 and Monkeypox virus lineage database updates.
- Mycobacterium tuberculosis antimicrobial resistance database update to WHO v2. Pretomanid resistance prediction is newly reported.
- Relative abundance and reads per million (RPM) as reported in the per-sample reports are now with a denominator that includes host reads. Previously, the denominator was the number of reads after host removal. For example, if 50/100 reads were human and 1/100 E. coli, the previous relative abundance for E. coli would have been 1/50 and it’s now 1/100
- More accurate metagenomic classification by handling classifications which result from low complexity regions.
- Improved pathogenicity prediction for eukaryotic organisms.
- Selective reporting of antimicrobials from rapidly growing Mycobacteria.
- Updated BugRef reference sequence database. Improved classification of sequences deriving from control sequences by masking contaminated reference sequences.
- Improved representation of Cryptosporidium species in BugRef.
Known issues at release¶
- Mean Phred scores per base in summary report are calculated as a simple average of all base Phred scores, instead of incorporating considerations for a logarithmic scale. Additional details are available in this blog post.
- AMR genes may be reported on a plasmid for drugs which aren’t included in the predicted phenotype panel.
- E. coli shiga toxin detection may miss multiple alleles if both occur in the same sample and data was generated with a Nanopore or PacBio sequencer.
5.3 - May 17, 2024¶
Added¶
- Antifungal resistance prediction for Candida auris.
- New columns and explanations to the plasmid tables in per-sample and summary reports:
- Subtype: plasmid subtype cluster IDs can be used to identify near identical plasmids and are more granular than cluster IDs.
- Predicted mobility: mobility prediction is based on the presence of relaxase, mate-pair formation and oriT sequences. A plasmid is classified as conjugative if it contains at least a relaxase and a mate-pair formation marker. Plasmids containing either a relaxase or an oriT but are missing the mate-pair formation marker are classified as mobilizable, while plasmids that are missing a relaxase and an oriT are classified as non-mobilizable.
- Mash distance to nearest NCBI reference sequence: see work by Robertson et al. for details.
- Detection and subtyping of C. botulinum toxin in appropriate Clostridium species.
- E. coli serotyping and shiga toxin detection.
- Report de novo assembly sequencing depth in the summary report for bacterial isolates. Note that this is slightly different than reference-based sequencing depth, which uses a species-specific reference genome to calculate coverage.
- Legionella pneumophila Sequence Based Typing (SBT).
- Streptococcus pneumoniae serotyping.
- Report multiple MLST schemes for organisms where there are multiple accepted schemes. If your organism of interest has multiple schemes but one or more aren’t reported, contact us to have it added.
- Output VCF files from Illumina analyses of viruses.
- Minority variant calling for M. tuberculosis resistance from Illumina data.
- Shigella genotyping.
- Report sequencing depth of genomes in the per-sample reports for Nanopore metagenomic samples.
- Report read duplication (column hidden by default) and remove duplicates for Illumina bacterial isolates.
Fixed¶
- Plasmid host range in the summary report plasmid overview table was previously blank.
- Cluster code generation which previously may have reported a close cluster code for genomes which were in fact distant.
Warning
This is a breaking change for cluster code addresses, as the address of old isolates may be updated upon a new analysis. BugSeq has contacted users reliant on cluster codes given the significance of this change.
- Bug affecting classification of contigs to the genus rank or above. Contigs originating from novel or under-represented species may have been unclassified. Benchmarking results and species-rank classifications are unchanged by this fix.
.fnaextension may have been shown in assembly completeness plots of per-sample reports.- Reference-based coverage calculation (in per-sample reports) was calculated based on PCR duplicate-marked alignments. However, duplicate marking was overly aggressive and coverage may have been underestimated.
- Improved detection of AMR genes that were broken at the ends of contigs. This resulted from gene annotation tools like Prokka and Bakta disabling gene calling at the ends of contigs.
Changed¶
- Improved accuracy for plasmid detection by using a curated repetitive sequence filter during plasmid identification. Previously, there may have been false negative detection of an IncX3 plasmid.
- Improved accuracy for plasmid detection by curating the plasmid reference sequence database. Excluded <0.02% sequences which are likely chromosomal.
- Revert: “Improved use of assembly graph for classifying Illumina contigs” in v5.2. In v5.2, there was an assumption that errors in the Illumina assembly graph were rare and the taxonomic bin could be assumed to be the same for an entire subgraph within the graph. After identifying several cases where graph edges connected nodes from unrelated taxa, we’ve reverted this change. BugSeq now uses a probabilistic framework to assess the edges between nodes in the assembly graph. This framework incorporates assembly graph edges, paired-read edges (if available) and neighbor labels. Subgraphs are no longer assumed to reflect the same taxon. Accuracy is improved over v5.2:
| v5.2 | Latest |
|---|---|
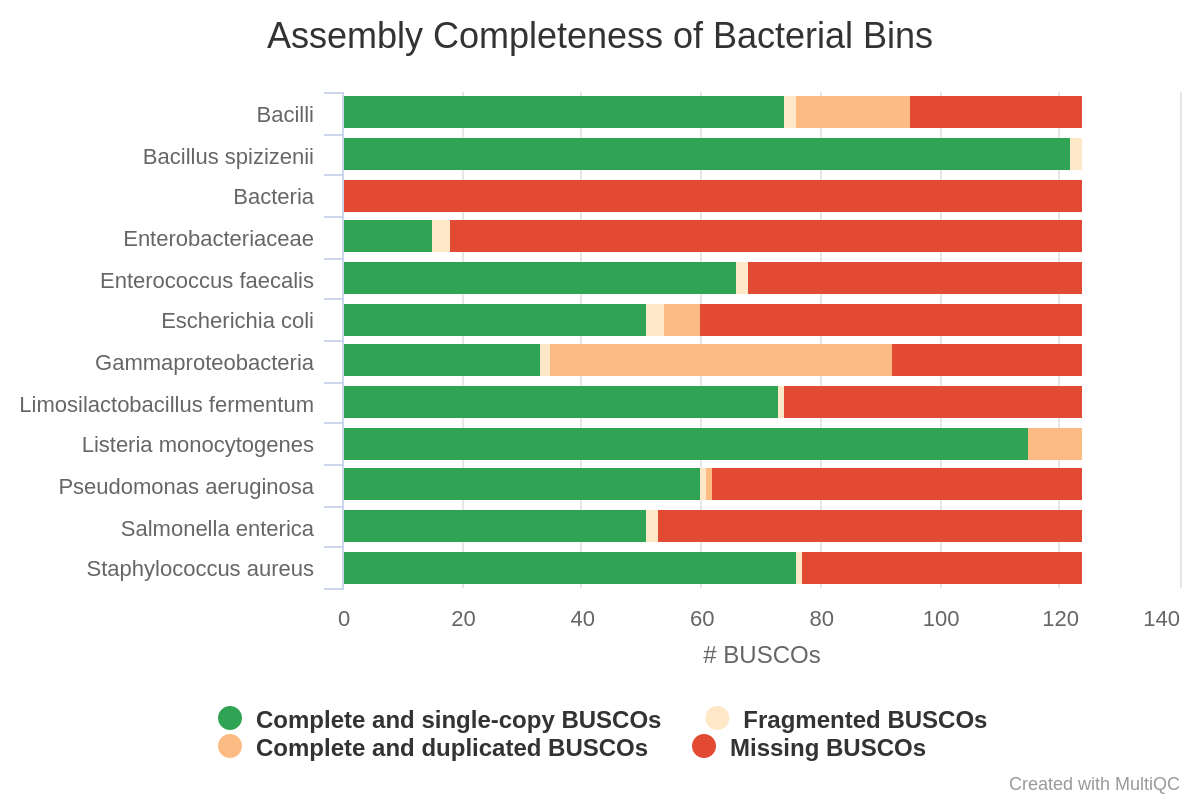 | 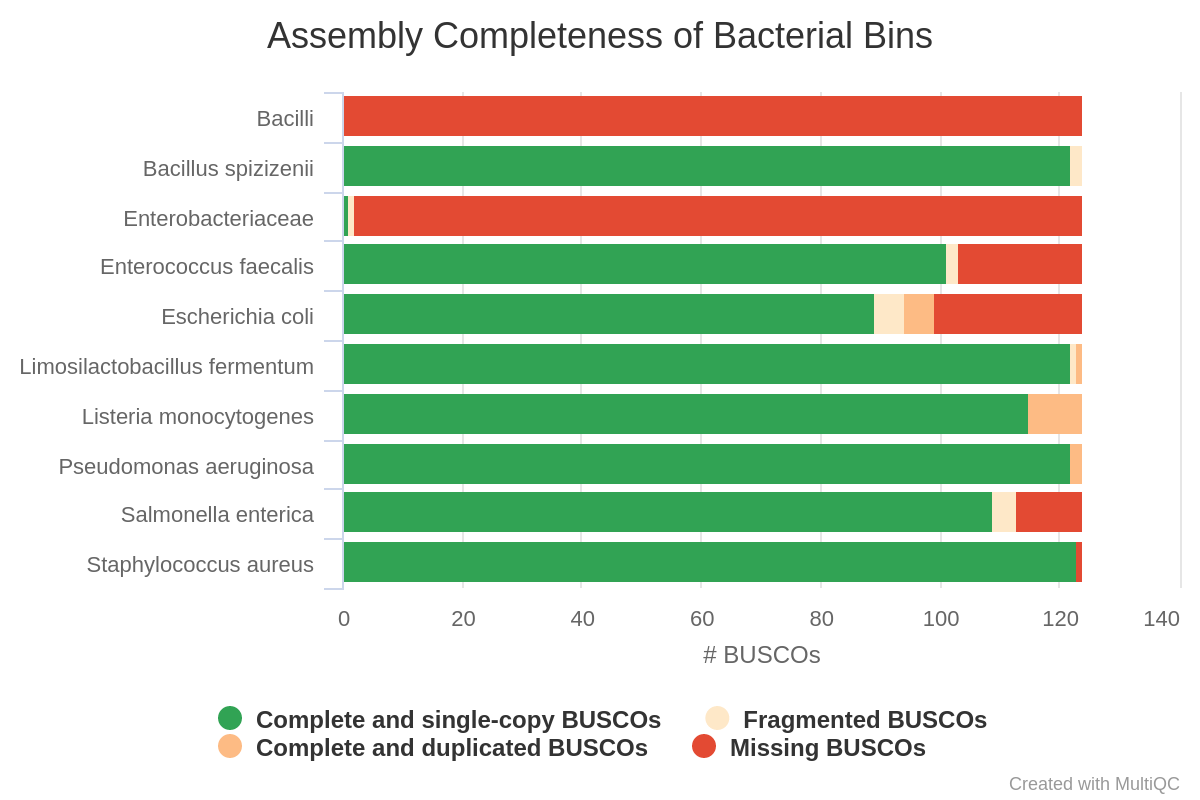 |
- Improved read trimming for Illumina and now enabled read trimming for other sequencing platforms. The improvement handles cases where there are very low quality bases at the ends of reads.
- Faster Nanopore assembly by using the new
lr:hqminimap2 preset for read-to-assembly alignment when appropriate. - Rename BugSeq Default DB to BugRef Curate DB in reports. See recent publication from our team on curation methods.
- FAST5 files are no longer accepted for upload. We recommend using ONT’s dorado for basecalling.
- Outbreak analysis
Distance MatrixandCluster Addresseshave been combined as separate sheets in a single excel output per-organism. A third sheet has been added with a distance matrix including only the samples of the viewed analysis. - Disambiguate host and vector/control sequences in the summary report General Statistics table. This change enables users to better understand why reads were filtered from the sample before metagenomic classification.
- Improved visualization of refMLST outbreak clusters.
- Improved recovery of contigs from repetitive regions when assembling short reads. The minimum contig length of short read assemblies has been decreased from 1000bp to a dynamically set threshold based on input read length.
- Further annotation of OXA-family beta-lactamases.
- Improved classification of Clostridium haemolyticum and Clostridium novyi.
- Updated reference sequence database. This update bring prophage masking, which should yield improved classifications for reads deriving from phages. It also introduces additional representation of Influenza A and Morganella species for improved detection.
- Classify more Illumina reads to deeper ranks by performing a two step classification including a profiling step and then a binning step.
- Classify viruses to more commonly known names, such as Influenza A virus instead of Alphainfluenzavirus influenzae.
- Improved host filtering of Nanopore and PacBio reads by incorporation of base quality into the filtering algorithm. Host filtering is more specific because it won’t filter reads which are divergent from human yet accurate.
- Dynamic minimum alignment length for Nanopore read-based classification. This change improves specificity; there should now be fewer false positive species detected at very low abundance.
- Rotate circular contigs for better detection of AMR genes at the ends of contigs.
- Update
pangolin-datato 1.27.
Known issues at release¶
- Mean Phred scores per base in summary report are calculated as a simple average of all base Phred scores, instead of incorporating considerations for a logarithmic scale. Additional details are available in this blog post.
- AMR genes may be reported on a plasmid for drugs which aren’t included in the predicted phenotype panel.
- E. coli shiga toxin detection may miss multiple alleles if both occur in the same sample.
- Running the same sample through BugSeq multiple times on the same analysis version may rarely result in different plasmids detected. A fix has been submitted to the upstream tool.
Median Read LengthandAverage Read Lengthcolumns (Group:Read Quality Control) reported in hidden columns in the General Statistics table of the summary report are inaccurate. These columns must be explicitly unhidden viaConfigure columnsto view the data. This error results from MultiQC inaccurately calculating these statistics from FastQC outputs. Please use theMedian Read Lengthcolumn shown by default, which is accurately calculated by a custom BugSeq tool.
5.2 - Feb 8, 2024¶
Added¶
- Influenza analysis enabling typing of Influenza A using amplicon or metagenomic approaches.
- Quality control note to accompany quality control flag of Pangolin SARS-CoV-2 variant calling (see MultiQC PR #2157)
- Hepatitis B virus genotyping reported on summary and per-sample reports from nanopore amplicon sequencing data.
- Output assemblies have contig length and plasmid name (if they’re classified as a plasmid) reported in sequence headers. The reported format is:
>contig1 len=4
AGTC
>contig2 len=4 plasmid=AA800
CTGA
- Illumina barcode crosstalk correction based on a method we described in Nanopore metagenomic sequencing for detection and characterization of SARS-CoV-2 in clinical samples.
Warning
Barcode crosstalk correction for Illumina is based on a general approximation of the crosstalk rate seen across Illumina datasets. The observed barcode crosstalk rate depends on many factors, such as the use of dual-indexed versus single-indexed adapters. For customization to your experimental design, get in touch.
Required data for application of crosstalk correction
Barcode crosstalk correction for Illumina relies on detection of the sequencing run ID in read headers of the FASTQ files. If the sequencing run ID isn’t found (eg. files downloaded from SRA), crosstalk correction won’t be applied.
- Confidence reporting for the absence of antimicrobial resistance. As detailed in reports, high confidence reflects that BugSeq obtained a complete or near complete genome for the organism and therefore judges it unlikely that there are factors predicting AMR which were missed. Conversely, incomplete genomes have lower confidence because predictors of resistance may have been missed.
- Visualization of refMLST outbreak analysis results with a minimum spanning tree.
- Generate consensus sequences for all viruses from Illumina sequencing data.
- Hidden columns to General Statistics table of per-sample report with additional details on each taxon (eg. rank and NCBI Taxonomy identifier).
Changed¶
- Minimum contig length for DNA analyses is set to 1000bp and for RNA/TNA analyses is dynamically set based on read length. This change significantly improves assembly quality and binning.
- New ensemble metagenomic classifier incorporating the results of multiple metagenomic classifiers. This is our largest upgrade for metagenomics since Version 2.0. Performance is improved for low abundance pathogens while maintaining leading precision. Notably, there are no longer results for both the read-based and assembly-based classifiers, simplifying outputs and interpretation of results.
- Improved nanopore RNA assembly accuracy. This change improves the detection of known and novel viruses using nanopore RNA-Seq.
- Update pangolin database for SARS-CoV-2 lineage calling.
- Plasmid database update which improves host range prediction.
- Update curated reference sequence database, bringing:
- Improved representation of eukaryotes, including fungi and protozoa.
- Improved filtration of genomes which are taxonomically misidentified.
- Hide Hepatitis B virus variants which derive from genotypic variation.
- Improved quality control of Legionella serogrouping if serogroup 14 is detected, which now reports a QC failure.
- Output assemblies are sorted from largest to smallest contig.
- Polishing of short read isolate assemblies resulting in higher base accuracy.
Note
Allele distances from refMLST may be reduced based on this change.
- BugSeq no longer filters reads from animal species (eg. mouse, rat, pig, etc.) before processing by default. These genomes were found to be contaminated with microbial sequences and filtering impacted downstream analyses. Human reads continue to be filtered by default.
Processing metagenomic data from animals
If you are performing metagenomic sequencing of an animal, get in touch with BugSeq before data submission for optimal host read filtration.
- Improved classification of contigs by masking AMR genes before alignment against the reference database.
- Sequencing depth calculation along with depth distribution plot is now more granular. Previously, BugSeq used the depth of the de novo assembly contigs to predict sequencing depth of the reference genome. As depth of the de novo assembly was aggregated by contig, this process averaged out variation within each contig. With the update, BugSeq uses reads binned by taxonomy to calculate depth of the reference genome. This process yields maximal resolution for plotting and investigation.
- Scaffold Nanopore and PacBio assemblies for more contiguous assemblies.
- Improved use of assembly graph for classifying Illumina contigs.
- Selecting NCBI nt database with Illumina data uses nt for all stages of analysis, from taxonomic binning of assembly to classification of individual reads. Previously, Illumina analysis with NCBI nt relied on BugRef (default database) for read-level classification.
- Improved reporting of error messages for invalid FASTQ data. A line number where the error occurred is now reported if relevant.
- Output MycoSNP SNP distance matrix for Candida auris in Excel format for easier access.
- Percentage of host reads is now calculated after metagenomic classification and includes both the number of reads identified with the host read filter and the number of reads identified as host in metagenomic classification. The host read filter remains in preprocessing to protect privacy and increase accuracy.
- Improved identification of Enterovirus and other viral species by adjusting thresholds to determine known versus novel viruses.
- Leverage a chromosome sequence filter to reduce false positive plasmid detections in bacterial isolates.
Warning
This is a breaking change for those using plasmid detection for outbreak investigation. This change results in “selective depletion of plasmids that have integrated within specific lineages but are also autonomously replicating plasmids elsewhere.” Read more and an example use of this filter by Robertson et al (2023).
Fixed¶
Misassemblies,Mismatches/100kbpandIndels/100kpbcolumns were briefly displayed under Assembly Statistics table on the per-sample reports. These columns were previously hidden and are now restored to hidden by default. they’re hidden by default as the assembly is compared against the reference genome, so mismatches are often the result of strain variation. Details of this bug are available in MultiQC PR #2190.- PDF reports contained metagenomic classification plots for each taxonomic rank. This has been fixed to the previous behavior of only.
- Issues classifying novel species or those not found in the reference sequence database. Some bacterial isolates may have been overclassified to the nearest species.
- Read preprocessing was briefly reported on separate lines in the general statistics table.
- Bacterial isolate assembly cleaning (removing contigs thought to be from extraneous DNA based on coverage, taxonomic classification and assembly graph connections) was inappropriately applied to some users’ analyses. This may have resulted in removal of contigs from the assembly (less than 1% of total assembly length) containing important genes such as those predicting AMR. Analyses such as refMLST and MLST weren’t impacted.
- Improved taxonomic identification of C. botulinum and related species, for example, C. sporogenes, via curation of this section of the database to conform to Brunt et al taxonomy.
Known issues at release¶
- Mean Phred scores per base in summary report are calculated as a simple average of all base Phred scores, instead of incorporating considerations for a logarithmic scale. Additional details are available in this blog post.
- AMR genes may be reported on a plasmid for drugs which aren’t included in the predicted phenotype panel.
5.1 - Oct 20, 2023¶
Added¶
- Annotation of variants with drug fold change in CMV antiviral drug resistance analysis.
- Complete Illumina wastewater analysis, including SARS-CoV-2 variant detection, visualization and aggregation of lab data over time.
- Hepatitis B Virus nanopore analysis, including genotyping, variant calling and variant annotation.
- M. tuberculosis lineage typing.
- Improved quality control of pathogen-specific analyses such as Legionella serogrouping, H. influenzae serotyping, N. meningitidis serotyping, S. pyogenes emm typing, K. pneumoniae typing and more.
- MLST reporting in summary report.
Changed¶
- Completely new AMR prediction analysis. This overcomes several limitations and bugs in the custom ResFinder method previously used, as detailed in v5.0. Both precision and recall are improved across all bacteria, with particular improvements to novel allele detection. This update also brings many new species-level AMR models. A benchmark paper will be published on the new AMR analysis and database; we’re currently seeking academic partners interested in validation so reach out if interested.
- Data submitted as bacterial isolates but found to have low level abundance of additional microbes will lead to masking of these additional organisms from reports in order to clarify reporting.
- Improved low complexity filtering for Illumina reads, which leads to more precise taxonomic classification.
- Pathogen identification from metagenomic samples is now reported with a Likert scale of probability from “Very likely” to “Very unlikely”.
- Metagenomic and taxonomic database update.
- Improved deduplication of sequences in the metagenomic database, improving classification accuracy.
- Use of WHO and additional databases for Mycobacterium tuberculosis resistance prediction. Confidence score for TB AMR prediction is now based on WHO confidence. The database source of each variant is annotated in the sample report.
Fixed¶
- Bacterial isolate summaries may have failed to be generated in some bacterial isolate analyses.
5.0 - May 29, 2023¶
Added¶
- Barcode crosstalk correction for nanopore assembly-based metagenomic abundance calculation.
Warning
This change only affects abundance calculation and doesn’t affect assembly or taxonomic binning. Metagenomic bins for taxa deriving from crosstalk, even if corrected to 0% abundance, will still be output. A future update will hide metagenomic bins purely deriving from barcode crosstalk.
- Name of reference database used to all reports.
- Additional QC metrics included in bacterial isolate summary report, including mean read qualities, assembly N50, assembly L50, assembly duplication, and sequence identity to reference genome.
- Ability to opt out of outbreak analysis (which adds samples to permanent laboratory database). A check box is now available on the submission page to use this feature.
- Haemophilus influenzae and Neisseria meningitidis serotype prediction.
- Streptococcus pyogenes (Group A Streptococcus) emm typing.
- Reporting of localization of AMR genes to plasmids. See Known Issues at Release below for limitations around this reporting.
- Reporting of additional information on detected plasmids, including coverage, and replicon, relaxase and mate-pair formation typing.
- Reporting of percentage of input reads filtered with host read scrubber. This data is located in the general statistics table of the summary report.
- Some results from the per-sample reports are now available in aggregate form in the summary report. This includes:
- Streptococcus pyogenes emm typing
Fixed¶
- The median base quality, both within and across reads, is properly calculated in the summary report. Additional details are available in this blog post.
Warning
Median read Q score in the General Statistics was temporarily reported erroneously based on a median of all reads’ median error probability, instead of a median of all reads’ average error probability. See this GitHub issue for an example. This error tended to inflate the median read Q score statistic.
- Median read length in the General Statistics table of summary report is properly calculated.
- Zip file of summary results may have included a zip file of summary results.
- Capitalization of some taxon names.
- Clarified text in reports describing plasmid cluster IDs.
- Number of reads reported for Illumina analyses in general statistics table of summary report now accurately reflects the number of all reads passing quality control filters. Read counts may have been underestimated before this fix, as only a subset of reads were counted.
- Text now wraps inside cells of tables on per-sample and summary reports.
- Improved detection of AMR predictors from R10.4.1 nanopore sequencing data basecalled with the fast model.
Changed¶
- Assembly completeness in the bacterial isolate summary now reflects both single-copy and duplicated single-copy orthologs. The previous metric reflected only single-copy orthologs, and can be derived from the current metrics as
Assembly Completeness - Assembly Duplication = Unduplicated Assembly Completeness. For pure bacterial isolates, the difference should be less than 3% to Assembly Completeness. - Removed minimizer duplication heatmap from summary reports for Illumina read-based metagenomic classification. This plot didn’t add value to assessing false positive classifications. Future updates will integrate genome coverage metrics into Illumina read-based metagenomic classification for improved classification precision.
- Stringent demultiplexing is now enabled for nanopore sequencing data by default if submitted without barcoding data or if submitted as FAST5 files.
Note
Files submitted with barcode information in the filename or folder name (if a folder was uploaded) will be unaffected.
- Improved coloring of “Genome Completeness” visualizations to reflect the severity of missing, fragmented and duplicated single-copy orthologs.
- Speed optimizations for large Illumina metagenomic samples. Results shouldn’t be affected.
- Major AMR database updates. Improvements are made to multiple beta-lactamase groups, along with better consistency across drugs within the same class.
- Additional selective reporting of antimicrobials for specific taxa (eg. Enterobacter and Serratia spp.).
- New 16S analysis for Illumina sequencing data. Individual reads are error-corrected and classified against a reference database. This new analysis brings increased classification recall and precision on internal benchmarks. For users interested in diversity estimation, we advise to filter taxa with less than 0.1-1% abundance, or to analyze at the genus rank.
- Updated MLST database.
- Updated SARS-CoV-2 lineage database.
- Updated plasmid database for improved plasmid host prediction.
- New default reference sequence database. This update brings significant accuracy improvements to classification of sequences deriving from plasmids.
- Better visualization of detected genotypic markers in the summary report via heatmap-style table.
- Plot sequencing depth by amplicon for amplicon sequencing experiments.
- Novel plasmids are named by the closest sequence in NCBI. The novel plasmid name format is “Novel_
-like” plasmid. - Better precision for Illumina read-level classification.
- Improved host read filtering by building a more robust host reference database.
- Improved host read filtering for Illumina sequencing data, resulting in improved performance for bacterial isolates with regions of sequence similarity to host organism (eg. Neisseria gonorrhoeae).
- Improved quality control reporting for Salmonella serotyping by including descriptive warnings.
- Improved read quality control for nanopore datasets which weren’t multiplexed.
- Improved circularity detection for sequences in Illumina sequencing data.
- Remove adapter detection plot in summary report for non-Illumina data.
- Improved assembly of nanopore data by factoring in data characteristics into assembly process and optimizing assembler parameters. Assemblies should be equivalent or better (more contiguous with greater accuracy) across basecaller presets and flowcell chemistries.
- Faster Illumina assembly, with increased contiguity and accuracy from internal benchmarks. The new assembly process also results in reduced gene duplication in taxonomic bins.
Known issues at release¶
- AMR gene alleles may be incorrectly called if they have an abundance of silent mutations. The AMR gene family should still be correctly called.
- Multiple AMR genes may be called for the same genomic region/sequence if there is a tie to two or more nearest alleles for that sequence.
- AMR genes reported on plasmids may be discordant from AMR genes reported in the bacterial genome. Plasmid AMR gene detection currently relies on a different method from bacterial genome AMR gene detection, and these methods will be harmonized in a future update.
- Mean Phred scores per base in summary report are calculated as a simple average of all base Phred scores, instead of incorporating considerations for a logarithmic scale. Additional details are available in this blog post.
4.0 - November 7, 2022¶
Added¶
- QuAISAR-style coverage calculation available in the bacterial isolate summary spreadsheet.
- Conditional formatting of outbreak analysis distance matrices.
- Filter host and Phi X reads before Illumina assembly.
- Monkeypox consensus sequence generation and clade classification.
- Visualization of nanopore and Illumina read-level metagenomic classification results in summary reports.
Note
Illumina read-level metagenomic classification is currently experimental and functionality may change in the future.
- Barcode crosstalk correction for nanopore sequencing data. We follow our previously published algorithm described and validated by Gauthier et al. (2021).
- Report percentage of host reads in the general statistics table of the summary report for nanopore data.
Fixed¶
- BugSeq evaluated a new algorithm to generate our taxonomic classification database. After hearing from our users, this algorithm didn’t generate databases with the hoped performance and we’ve reverted the database change.
Warning
Analyses run between August 15 and September 6 may have been affected, and we encourage users to resubmit their data if analyzed during this period.
- Sort columns by sample name in summary AMR table.
- Erroneous blank line in some tables of PDF reports.
- Fixed cefixime reporting for ESBLs. Cefixime should now be flagged as having a genotypic predictor of resistance if an ESBL is present.
- Fixed ceftriaxone reporting for carbapenemases. Ceftriaxone should now be flagged as having a genotypic predictor of resistance if certain carbapenemases are present.
- Aggregate plasmid table reported a plasmid with name “0” as found when no plasmids were found.
- Barcode trimming is skipped if reads have already had barcodes trimmed.
- Bacterial isolate summary table is now sorted by sample name.
Changed¶
- All databases were updated. New database include broader taxonomic representation and should therefore provide increased classification accuracy.
- Outbreak analysis module can now handle isolates that have been submitted to BugSeq multiple times with the same name. Each time an isolate is submitted to BugSeq, we now record the date of submission to keep track of resequenced/duplicate isolates.
- Updated AMR database. Major updates are made to OXA-type beta-lactamases, which should more accurately represent the phenotype of individual families and alleles.
- Read count plot has been merged with read filtering plot in summary reports to reduce redundancy of results.
- Nanopore 16S analysis now accepts clusters of 30 reads or larger.
- Stringent demultiplexing for nanopore has temporarily been removed. BugSeq now relies upon the demultiplexing performed by the user. If you would like to analyze stringently demultiplexed data, please perform this before submitting to BugSeq.
- Speed optimizations.
- Keep reads with a greater number of Ns to discard less data.
- Improve nanopore RNA assembly speed and quality.
- Report median read length instead of mean read length in reports.
- Dynamically set contig length suffix (eg. bp, Kbp, Mbp) in reports.
- Filter host (eg. human) reads before metagenomic classification. This improves both speed (fewer reads need to be classified against the full database) and accuracy (some reads may have erroneously been classified to organisms with similar genomes to host).
3.0 - May 16, 2022¶
Added¶
- Flowcell/sequencing run quality control for nanopore sequencing data. If you submit FASTQ files to BugSeq containing sequencing information in the FASTQ headers, this will now be plotted by run ID on the summary report.
- Legionella serogroup prediction.
- Information on read filtering during preprocessing to summary reports for all nanopore sequencing experiments.
- Sample reports now contain additional strain typing information on Salmonella, Klebsiella and Legionella species.
- Bacterial isolate summary report in Excel format.
Fixed¶
- Better detection of nanopore 16S experiments by lowering the acceptable median length of 16S reads.
- Plot titles in reports now reflect the content of the plot instead of the tool used to generate them (which was sometimes erroneous).
- Fixed fraction of reference genome covered calculation for assemblies which were close to 95% sequence identity to the reference genome. In this scenario, reference genome coverage was vastly underestimated. Bacterial isolates with >99% sequence identity to the reference genome were unlikely to be affected. This issue is related to this bug in QUAST and BugSeq has implemented an internal fix pending an official fix on QUAST.
- refMLST allele calculation if there was a variant in the first or last base of a loci. Accounting for these variants increases resolutions and distance between isolates by an average of 2 alleles at distances less than 50.
- refMLST clustering of isolates that are equidistant to two separate clusters. Isolates meeting this criteria now cluster with the first cluster observed. Previously, their clustering was assigned randomly to one of the two or more equidistant clusters.
- Platform-specific thresholds for classifying sequencing quality control data as pass/warning/fail.
- Missing tables in PDF reports if they have too many columns. That’s now fixed with a message to check the HTML report for the full table.
Changed¶
- Summary reports now show total reads after filtering. This streamlines the summary reports for Illumina paired-end data as individual FASTQ files are no longer reported in the General Statistics table by default. Note that the number of reads after filtering is the sum of both paired-end files for Illumina.
- Krona plots for BugSplit (assembly-level) classification now shows all ranks, including intermediary ranks.
- Unclassified contigs (and the reads mapping to them) are now classified as unclassified instead of root in Kraken report formats.
- No longer expose unbinned assemblies. The unbinned assembly may be obtained by concatenating together all the binned assemblies.
- Removal of Racon/Medaka/Homopolish polishing of nanopore R9.4.1 and R10.3 assemblies for the following reasons:
- Unfortunately, both Racon and Homopolish had critical issues preventing their widespread use.
- On internal benchmarks, Medaka may decrease assembly quality if there is a mismatch between basecaller version and Medaka model.
- With recent improvements in nanopore basecalling accuracy, polishing no longer has a significant impact on assembly quality.
- Polishing was adding to processing time but wasn’t increasing metagenomic classification or downstream analysis accuracy (eg. AMR). BUSCO completion analysis may be impacted with more fragmented genes detected; however, this doesn’t impact other analyses.
- Faster Illumina analyses by allocating more CPUs to the assembly process.
- don’t bin long-read assembled contigs less than 1000bp in length. This increases accuracy of analysis as these contigs were the most likely to be erroneously classified, and should also not be present in long-read assemblies.
- Expose additional antibiotics on sample reports for Mycobacterium tuberculosis.
- Allow IUPAC characters in input sequencing data.
- Improved taxonomic binning of bacterial isolates.
- Updated AMR and MLST database.
- Bin plasmids to their host bacteria across all sample types.
2.3 - January 25, 2022¶
Added¶
- Include the following data in individual sample reports:
- Antimicrobial resistance table
- TB spoligotyping
- Sample name
- Table of plasmids detected
- Canonical SNP typing for:
- B. anthracis
- F. tularensis
- Y. pestis
- C. burnetii
- Include the following data in aggregate reports:
- Plasmids detected across all samples
- Antimicrobial resistance detection across all samples
- Read filtering statistics
- PDF generation of reports
- Assembly and processing of Q20+ ONT sequencing data
- Insertion and deletion detection for Bacillus anthracis
- cDNA amplification primer trimming on metatranscriptomic nanopore data
Fixed¶
- Error handling if a user submits duplicated reads across multiple files
- Coverage per contig plot in sample reports
- Reporting of first line antimicrobials for Mycobacterium tuberculosis
Changed¶
- Improved detection of plasmid sequences
- Improved assembly of ONT cDNA/RNA-sequencing data
- Improved abundance calculation from Illumina assemblies
- Improved primer scheme detection and trimming for SARS-CoV-2 amplicon sequencing data
- Faster nanopore 16S processing by setting the QIIME2 vsearch
--maxrejectsflag. On internal evaluations, this has negligible impact on results but drastically speeds up processing. - Faster overall analyses by being more selective in which pathogen-specific analyses get run
- Gentler quality filtering for Illumina data should result in better and more contiguous assemblies
- Faster read preprocessing by better leveraging parallel processing
- Removed raw text file output of AMR data as it’s now contained in reports and much easier to use
2.2 - October 26, 2021¶
Added¶
- Plot BUSCO results in reports
- Include MLST results in per-sample report
- Report analysis pipeline version in reports
Fixed¶
- Bug handling many Illumina samples in one analysis
- Depth calculations only included reference contigs with mapping assembled contigs. This is now fixed - sequencing depth may be slightly lower than previously reported.
Changed¶
- BugSplit can now report strain-level classifications
- Upgraded
seqkitfor faster FASTQ parsing
2.1 - October 14, 2021¶
Added¶
- Per-sample HTML report
- Sequencing depth calculation performed for each genome in a sample relative to its reference sequence
- Stringent demultiplexing of user submissions with custom file names
- Visualization of Pangolin SARS-CoV-2 lineage results in the summary report
Fixed¶
Changed¶
- Improved taxonomic binning of Illumina sequences using assembly graph information
- Improved Illumina assembly by merging paired-end reads
- Faster internal file transfers, resulting in faster analyses
2.0 - October 7, 2021¶
Added¶
- Assembly and polishing of data from all sequencers
- BugSplit module: high-accuracy taxonomic binning (Citation)
Fixed¶
- Respect a user’s custom filename
Changed¶
- Only demultiplex nanopore if it doesn’t have a custom filename
1.0.0 - December 3, 2019¶
Added¶
- First release!