Input¶
Uploading Data¶
BugSeq has two options for submitting data to the platform. All data is stored securely and encrypted, and is only accessible to your own account.
Uploading Directly¶
To upload files from your computer/server, drag and drop all files into the upload box, or select your files by clicking on the upload box.
Tip
Submit all files for your analysis together (ie. multiple samples) to speed up processing time and perform cross-sample analyses.
BaseSpace Integration¶
For BaseSpace users, we have an integration to retrieve data directly from BaseSpace. This is significantly easier, faster and less error-prone.
Once it is set up, your projects will appear in BugSeq. When you click on a BaseSpace project, you will see the samples in the project and have the ability to add them to an analysis:
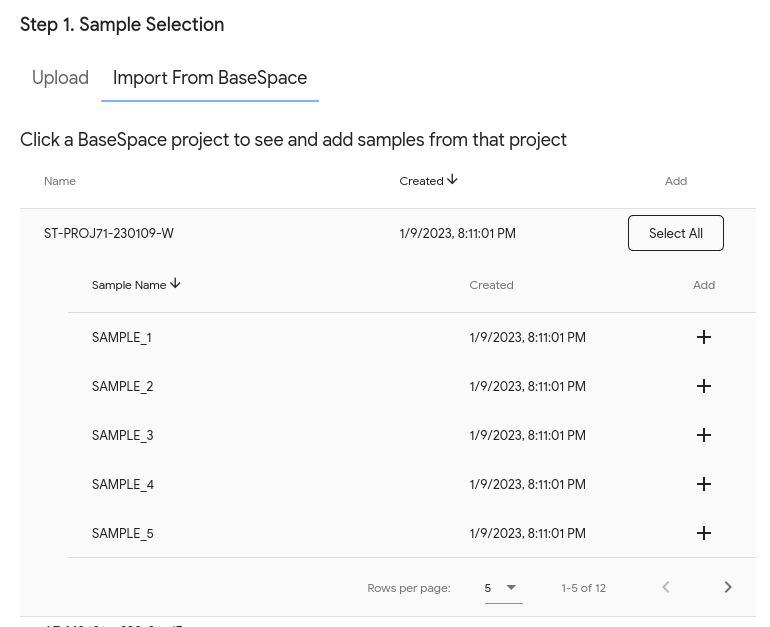
We require your BaseSpace API key to link BugSeq with BaseSpace. API keys are stored securely and encrypted within the BugSeq infrastructure, and are not accessible to anyone.
To get onboarded to our BaseSpace integration:
- Please reach out to support@bugseq.com so we can securely obtain your BaseSpace API key.
- We will enable the integration on your account.
- We will confirm once this is complete by responding to your email.
- You’re ready to go!
Optimizing Nanopore Output for BugSeq¶
To achieve maximal upload and runtime performance, prefer larger per-barcode files over thousands of small files.
The number of reads per FASTQ file can be specified in MinKNOW GUI when configuring the sequencing run. At the final step where output format is specified, users can modify the number of reads (records) per FASTQ file. For the most efficient analysis speed, we recommend selecting a value of at least 100,000, and possibly as high as 500,000 (much larger than the default of 4,000 reads per FASTQ file).
Modifying MinKNOW settings is best as the resulting files will still conform to ONT’s file naming conventions, which BugSeq can parse. You can also concatenate FASTQ files manually for Oxford Nanopore sequence data into a smaller number of files per sample. However, please be careful to adhere to BugSeq’s input filename requirements.
Format¶
BugSeq supports all standard sequencing file formats. The following file formats are currently accepted:
FASTQ: Basecalled nanopore, PacBio or Illumina sequences- Acceptable file extensions:
- Uncompressed:
.fq,.fastq - Compressed:
.fq.gz,.fastq.gz
- Uncompressed:
- Note: only DNA/RNA sequencing data with IUPAC notation is acceptable.
- Acceptable file extensions:
BAM: Basecalled nanopore or PacBio sequences- Acceptable file extensions:
.bam - Note: all reads must be unmapped, such as those directly output by the sequencer or basecalling software. BAM files with mapped reads will fail analysis.
- Acceptable file extensions:
Info
We accept both single and paired-end Illumina data. Paired-end data must contain separate files for forward and reverse reads with format {SAMPLE_NAME}_R[1/2].fastq[.gz], {SAMPLE_NAME}_[1/2].fastq[.gz] or {SAMPLE_NAME}_L[/d]+_R[1/2]_001.fastq[.gz].
Tip
Uploading gzip compressed files will reduce transfer time of large files to BugSeq.
Metadata Submission¶
Metadata can be submitted alongside sequencing data and is incorporated into various parts of our platform. Examples of annotating samples and the subsequent impact include:
- Samples can be specified as a negative control. Read counts in other samples will be normalized against the read counts in the negative control.
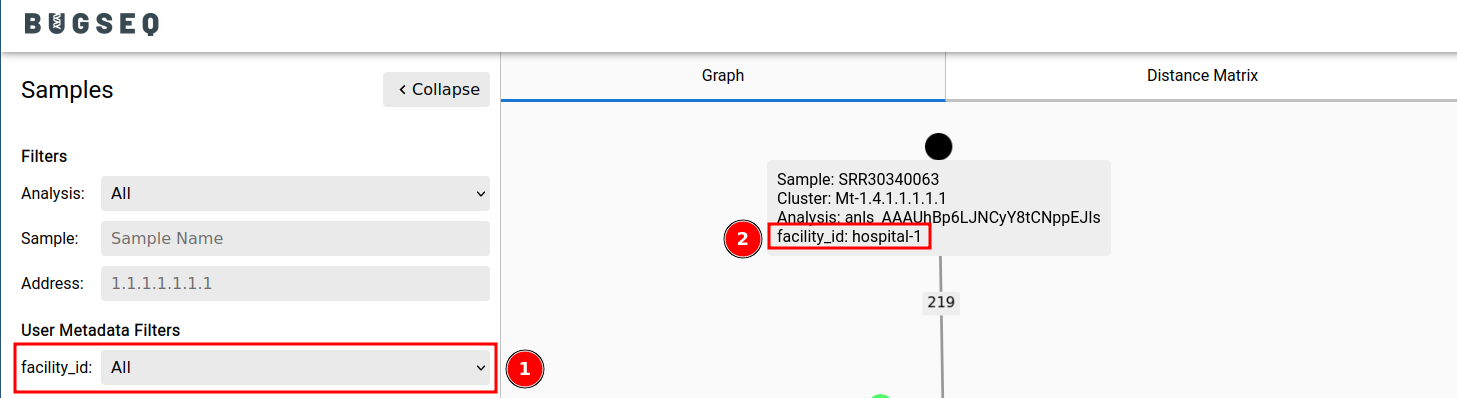
- Samples can be annotated with arbitrary fields. For example, a sample can be annotated with
facility_id: hospital-1. The metadata will appear in outbreak analyses.-
Example of Metadata Incorporated into Outbreak Analysis
-
Info
Metadata must be enabled for your account before you can submit metadata. Please get in touch if you would like to make use of metadata.
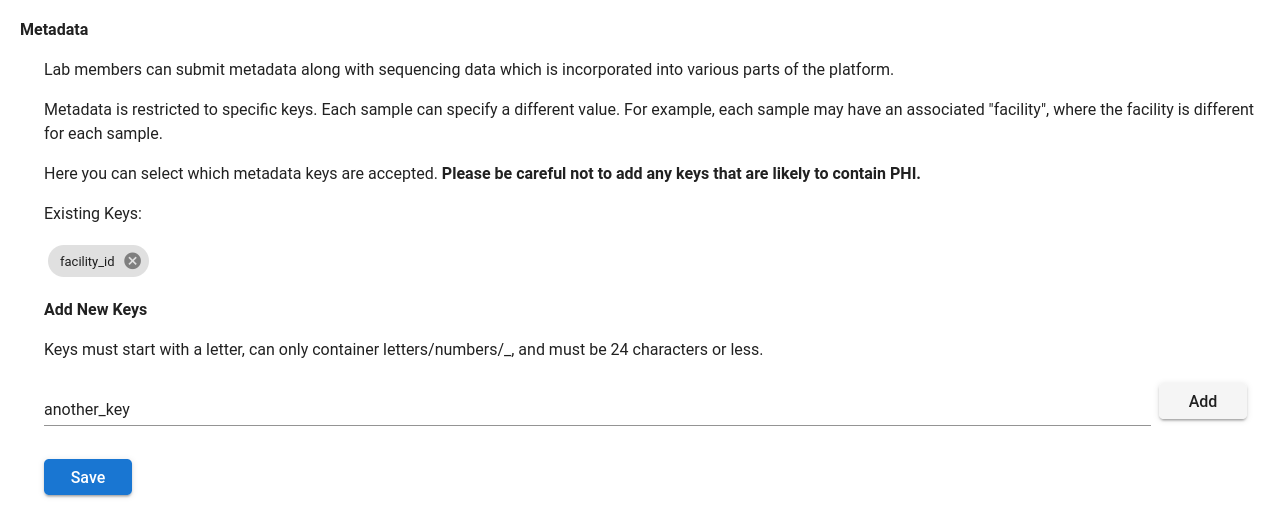
Specifying Metadata Keys for a Lab¶
In order to annotate a sample with metadata, a lab manager must add the key to the lab as an eligible metadata key. Metadata keys can be configured under “Labs”, on the “Data Access & Notifications” tab. Here you can add or remove keys.
Please be careful not to add any keys that are likely to contain protected health information.
Annotating a Sample with Metadata¶
We recommend uploading files first, as the upload form will help annotate recognized samples.
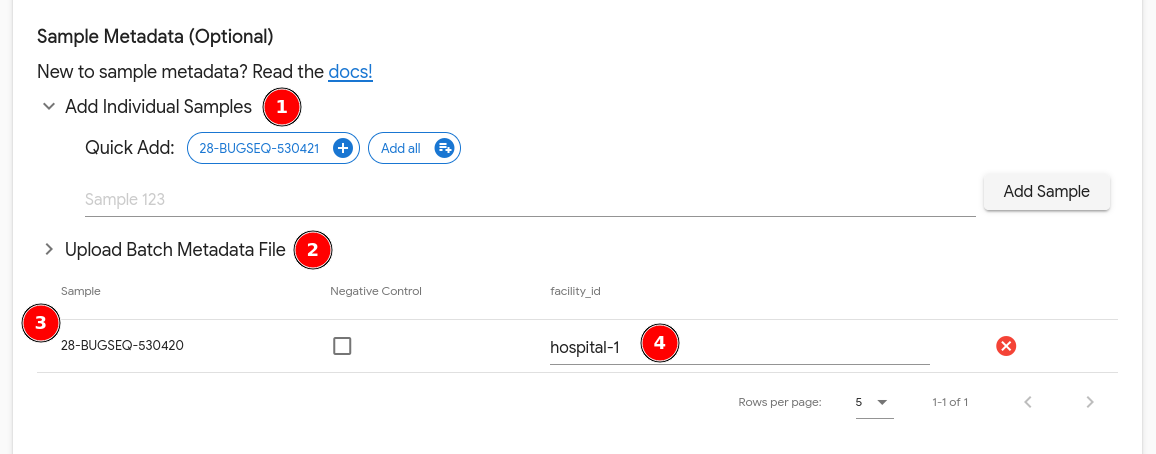
Samples can be added in 2 ways:
- Add each sample individually. If sample names can be detected from input files, click on the “Quick Add” samples to add them directly. Otherwise you can manually input a sample name and click “Add Sample”.
-
Add samples in bulk using a CSV file. The permitted columns are
sample(the name of the sample),negative_control(values can betrueorfalse) and any custom metadata keys enabled on the lab that the analysis is being submitted under. We recommend downloading the provided template and filling it out in Excel.
Not all submitted samples need to be annotated with metadata.
Warning
All samples that are annotated must have matching sequencing data submitted. If metadata is submitted for a sample that does not have any sequencing data, the analysis will fail.
Read Preprocessing¶
As BugSeq performs automatic read preprocessing and leverages all aspects of the input data to produce optimal results, we recommend against any data manipulation before submitting to BugSeq. For example, BugSeq uses FASTQ quality information to correct reads, yielding optimal assemblies. BugSeq is built to handle raw data, saving our users time and complexity before data submission while ensuring optimal results.
Experimental Design¶
Platform¶
You may submit files from any Oxford Nanopore, PacBio or Illumina sequencer.
BugSeq will automatically detect the sequencing platform and perform tailored, best-practice analyses. The following processes are adjusted based on platform:
- Basecalling
- Demultiplexing
- Quality evaluation
- Adapter trimming
- Quality trimming and filtering
- Metagenomic classification
- Alignment
- Assembly
- Taxonomic binning
- Post-assembly analyses, including:
- Antimicrobial resistance prediction
- Strain typing
- Other pathogen-specific analyses
Sequencing Strategy¶
BugSeq supports all widely accepted sequencing strategies. These include:
- Metagenomic/Metatranscriptomic Sequencing
- Whole Genome Sequencing (WGS)
- Amplicon Sequencing
- 16S/ITS
- MLST
- Viral amplicons
Sequenced material can be DNA or RNA. Nucleic acid can be sequenced directly or amplified with techniques such as PCR.
Multiplexing¶
BugSeq automatically performs demultiplexing and adapter trimming on nanopore sequencing data. Illumina and PacBio data should already be demultiplexed before submission, but may have adapters, as BugSeq trims these automatically.
Strict (dual-barcode) nanopore demultiplexing
BugSeq parses FASTQ headers for barcoding data. Occasionally, users may want BugSeq to perform strict demultiplexing of nanopore data, looking for barcodes on both ends of reads. Users should either perform strict demulitplexing before submitting to BugSeq, or perform no demultiplexing before submitting to BugSeq. Files which have already been demultiplexed with default (single-ended barcode) demultiplexing will not be further demultiplexed by BugSeq.
New Designs¶
Don’t see your experimental design combination here? Get in touch with us via support.