Data submission¶
Uploading data¶
BugSeq has two options for submitting data to the platform. All data is stored securely and encrypted, and is only accessible to your own account.
Uploading directly¶
To upload files from your computer/server, drag all files into the upload box, or select your files by clicking on the upload box.
Tip
Submit all files for your analysis together (ie. multiple samples) to speed up processing time and perform cross-sample analyses.
BaseSpace integration¶
For BaseSpace users, BugSeq has an integration to retrieve data directly from BaseSpace. This is significantly easier, faster and less error-prone.
Once it’s set up, your projects will appear in BugSeq. When you click on a BaseSpace project, you will see the samples in the project and have the ability to add them to an analysis:
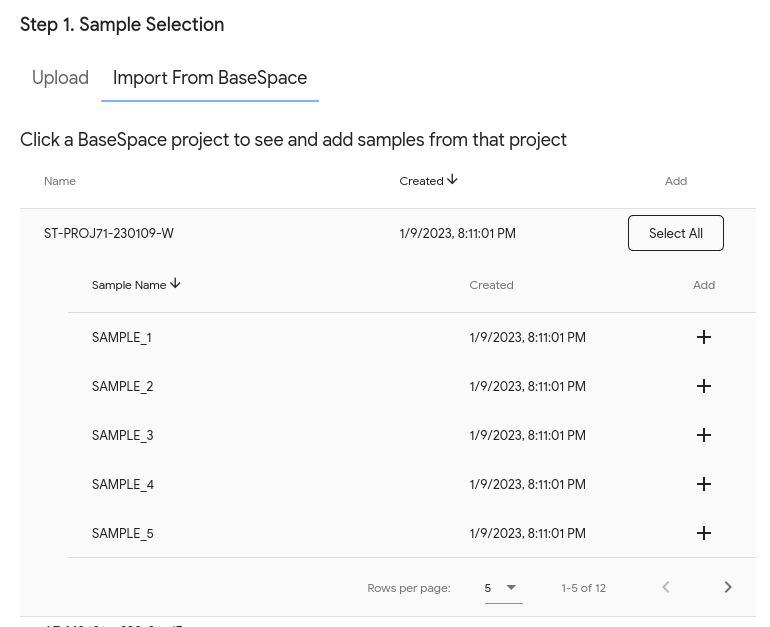
We require your BaseSpace API key to link BugSeq with BaseSpace. API keys are stored securely and encrypted within the BugSeq infrastructure, and aren’t accessible to anyone.
To get onboarded to our BaseSpace integration:
- Please reach out to support@bugseq.com so we can securely obtain your BaseSpace API key.
- We will enable the integration on your account.
- We will confirm once this is complete by responding to your email.
- You’re ready to go!
File format¶
BugSeq supports all standard sequencing file formats. The following file formats are currently accepted:
Info
We accept both single and paired-end Illumina data. Paired-end data must contain separate files for forward and reverse reads with format {SAMPLE_NAME}_R[1/2].fastq[.gz], {SAMPLE_NAME}_[1/2].fastq[.gz] or {SAMPLE_NAME}_L[/d]+_R[1/2]_001.fastq[.gz].
FASTQ: Basecalled ONT, PacBio, Ion Torrent or Illumina sequences- Acceptable file extensions:
- Uncompressed:
.fq,.fastq - Compressed :
.fq.gz,.fastq.gz
- Uncompressed:
- Only DNA/RNA sequencing data with IUPAC notation is acceptable.
- Acceptable file extensions:
BAM: Basecalled nanopore or PacBio sequences- Acceptable file extensions:
.bam - BAM files must be already demultiplexed and split into one file per barcode.
- All reads must be unmapped, such as those directly output by the sequencer or basecalling software. BAM files with mapped reads will fail analysis.
- Acceptable file extensions:
Tip
Looking for details on how samples get named in BugSeq reports? See the demultiplexing section.
Read preprocessing¶
As BugSeq performs automatic read preprocessing and leverages all aspects of the input data to produce optimal results, we recommend against any data manipulation before submitting to BugSeq. For example, BugSeq uses FASTQ quality information to correct reads, yielding optimal assemblies. BugSeq is built to handle raw data, saving our users time and complexity before data submission while ensuring optimal results.
Experimental design¶
Platform¶
You may submit files from any Oxford Nanopore, PacBio or Illumina sequencer.
BugSeq will automatically detect the sequencing platform and perform tailored, best-practice analyses. The following processes are adjusted based on platform:
- Basecalling
- Demultiplexing
- Quality evaluation
- Adapter trimming
- Quality trimming and filtering
- Metagenomic classification
- Alignment
- Assembly
- Taxonomic binning
- Post-assembly analyses, including:
- Antimicrobial resistance prediction
- Strain typing
- Other pathogen-specific analyses
Sample type¶
BugSeq supports a broad range of sample types, from clinical specimens to environmental samples. The selected sample type will impact certain aspects of the analysis pipeline, reflecting the results that you will see in the results page.
Tip
We recommend performing separate analyses for each sample type so that results are optimized for the input data.
Sample type selection can impact the following BugSeq outputs:
- Pathogenicity prediction: BugSeq maintains a comprehensive list of known and probable pathogens specific to certain body sites, which are reflected in the metagenomic classification table in Per-Sample Reports. We don’t perform pathogenicity prediction if “Generic” is selected as the Sample Type.
- Isolate Summary (Per-Sample Reports): Isolate identity and summary information is only provided if “Isolate” is selected as the Sample Type.
- Isolate Summary (Other Reports): Found under “Other” in the result page, BugSeq outputs an Excel-formatted (
.xlsx) isolate summary file for all samples submitted under a given analysis when “Isolate” is selected as the Sample Type, which includes QC statistics, as well as information on AMR genes and plasmids for each sample.
Not seeing an expected output?
If you don’t see a result that you expect based on your desired analysis parameters, please see the Output page for more details that may impact the results, or get in touch with us via support if you have any questions.
Sequenced material¶
In the upload form, you can specify DNA, RNA, or both (Total Nucleic Acid; TNA) as the Sequenced Material. This impacts how results are ordered in the Per-Sample reports for metagenomic classification. For example, if DNA is selected as the Sequenced Material, viruses with only an RNA stage in their lifecycle will be flagged as potential contaminants and moved to the bottom of the classification table, triggering a text warning at the top of the report.
Selecting the correct Sequenced Material is also important for optimizing the performance of our analysis, as different tools are used for assembly and classification depending on whether DNA or RNA/TNA is selected as the Sequenced Material.
Sequencing strategy¶
BugSeq supports all widely accepted sequencing strategies. These include:
- Metagenomic/Metatranscriptomic Sequencing
- Whole Genome Sequencing (WGS)
- Amplicon Sequencing
- 16S/ITS
- MLST
- Viral amplicons
Nucleic acid can be sequenced directly or amplified with techniques such as PCR.
New designs¶
Don’t see your experimental design combination here? Get in touch with us via support.
Lab selection¶
Labs are a great way to help organize your data in BugSeq; see the Labs section of our docs for more details on how to use, customize, and configure Labs.